

WS2 案例理论研究:通过相变提高储氢性能
摘要
氢是一种高效的清洁能源,但其储存和运输问题仍然阻碍其广泛使用。由于大的比表面积和独特的电子结构,二维材料在储氢方面具有巨大的潜力。特别是,单层 2H-WS2 已被证明适用于储氢。但关于 WS2 的其他两相(1T、1T')在储氢中的研究很少。在这里,我们进行了第一性原理计算来研究 WS2 的所有三相的氢吸附行为。多项氢吸附研究也评估了这些材料的储氢能力。综合分析结果表明,1T'-WS2比2H-WS2具有更好的储氢性能,这意味着相工程可能是提高储氢性能的有效途径。该论文为进一步研究二维材料储氢提供了参考。
介绍
传统的储氢由于其低燃点、广泛的可燃性和钢脆化而具有相当大的风险 [1, 2]。虽然金属氢化物(如 CaH2)可以储存大量氢气,但它们不仅在潮湿时易燃,而且价格昂贵且难以重复使用。因此,寻找一种安全、经济、有效的储氢材料已成为人们普遍关注的问题[3]。由于其较大的比表面积和独特的电子性质,二维(2D)材料已广泛应用于光催化分解水、析氢反应、晶体管、电致发光器件、储气和气体吸附等许多领域[4, 5,6,7,8,9]。例如,吸附在石墨烯上的氢需要通过转换 C–C π 来重新杂化碳价轨道 键与 C–H σ 键,这可以在费米能级周围引起带隙和磁矩,因此石墨烯的氢化提供了一种令人兴奋的可能性,可以直接在原子尺度上用预先设计的图案编写电子电路 [10]。石墨烯基材料的成功也推动了对应用于气体吸附或存储的其他二维材料的研究 [11,12,13,14]。更重要的是,单层过渡金属二硫化物(TMD)材料尤其表现出优异的储氢性能[15]。
储氢能力可以通过气体分子在材料表面的吸附强度来评估[16]。吸附强度不宜过强或过弱,因为目标气体分子在强吸附力下难以与材料分离或在弱吸附力下不稳定吸附[17]。在室温(约 25 °C)下,合适的储氢材料的每个氢分子的平均结合能为 - 0.2 至 - 0.6 eV [12]。然而,石墨烯或 TMD 等原始材料的不足之处在于它们与氢分子的结合力太弱 [18, 19]。通常采用表面功能化方法来改善其吸氢性能。通过掺杂或装饰工艺,可以改变二维材料的表面特性以适应适度的氢吸附能范围,进一步提高储氢性能[20]。然而,很难保持装饰系统的稳定性[21, 22]。并且准确地掺杂或装饰具有挑战性[23]。这种方法理论上可行,但远未应用。
作为典型的 TMD,MoS2 和 WS2 已证明其在储氢领域具有出色的应用潜力 [24, 25]。由于其优越的催化性能和独特的电学性能,MoS2在许多领域受到广泛关注[26],而WS2却经常被忽视。与单层 MoS2 相比,WS2 具有更好的热稳定性 [27, 28] 和在压缩应变下与氢分子的更大结合能 [29]。众所周知,WS2 还具有另外两个相(1T/1T'),它们具有不同的对称性和不同的电子特性。先前的研究表明,它们可以通过简单的方法制备 [30, 31]。大多数方法基于从 2H 相 WS2 的相变并结合稳定方法。许多研究表明成功制备了高百分比和稳定的 1T/1T'-WS2(表 S1)。最近,金属 1T-WS2 及其衍生物 1T'-WS2 在析氢反应 (HER) 应用中显示出巨大的潜力 [23, 32]。研究结果表明,它们的表面对反应中间体H*具有中等吸附强度。这为他们与氢吸附相关的其他应用(如储氢)铺平了道路。然而,关于这两个 WS2 相的储氢特性的研究很少。相位差对储氢的影响一直被忽视。
在这项工作中,我们研究了 WS2 的所有三个相,以比较它们作为储氢材料的适用性。我们对结构进行了系统的理论研究,并分析了气体分子的吸附能和吸附构型。为了模拟真实的工作条件,研究了大量氢分子的吸附。根据这项工作的计算结果,我们发现 1T'-WS2 是这三相 WS2 中作为储氢材料的最佳候选。改变 WS2 的相可以改善储氢。从而为二维材料的相学储氢研究提供参考。
计算细节
基于密度泛函理论(DFT)使用第一性原理。这项工作中的所有计算都是在 Dmol3 [33] 中进行的。局部密度近似 (LDA) 用于通过 PWC 函数处理交换和相关势。单一生产潜力被用来代替内核(DFT 半核赝品)以降低计算成本。通过选择双数值轨道基组和轨道极化函数 (DNP) 实现了更高的精度。然后,进行了收敛测试。经过测试,Monkhorst-Pack k -points 设置为 4 × 4 × 1,然后制作 20 Å 的真空层以防止层间相互作用。能量收敛精度设置为1×10 −5 Hartree (1 Hartree =27.212 eV),最大位移为0.005 Å,原子力不超过0.002 Hartree/Å。后面的所有计算都遵循这些属性。
对于这三相 WS2(1T/1T'/2H),计算模型为 4×4 单层 WS2 的超胞。 1T-WS2和2H-WS2结构首先由我们自己建造。施工完成后,进行几何优化,包括单元优化。 1T'-WS2 是基于现有的 1T'-MoS2 构建的。虽然 1T'-MoS2 是基于 2×2 1T 模型构建的,但单个氢原子被设置为 1T MoS2 的 S 原子的关键连接。然后,对系统进行了另一次几何优化。优化后,去除氢原子并再次优化以获得规则的1T'-MoS2结构。之后,在一个2×2的模型中,所有的Mo原子都被W原子取代,然后再进行几何优化,包括再次单元优化。
使用优化后的 2×2 WS2 模型,构建了 4×4 单层 WS2 的超胞。如图 S1 所示,WS2 的所有这三个相的模型在一个单元中包含 32 个 S 原子和 16 个 W 原子。由于 1T 相模型中呈现的 25 个 W 原子中有 16 个位于边缘或拐角处,因此晶胞中 W 原子的有效数量仍为 16 个。 1T 或 2H 模型中每个 W 原子之间的键相等,而1T'-WS2 中的不相等。由于 1T'-WS2 中的 W-W 键,W 原子的排列看起来像一个锯齿链。
因此,1T'相在一些研究中也称为锯齿链相。我们可以在具有共同特征的三种结构中找到重复单元。如图 1 所示,绿色框代表边缘只有 W 原子的重复单元,而红色框是由 S 原子勾勒的那些。由于对称性差异,1T'模型中绿框的大小几乎是1T模型中的两倍。 1T或1T'型号的红框是六边形,而2H型号则是三角形。 1T和1T'-WS2结构中也有类似的重复单元,如图S1中的蓝色矩形区域。此外,1T和1T'模型中红框所示的轴对称元素也可以在图S1中找到,也可以代表1T和1T'-WS2结构的对称性。
<图片>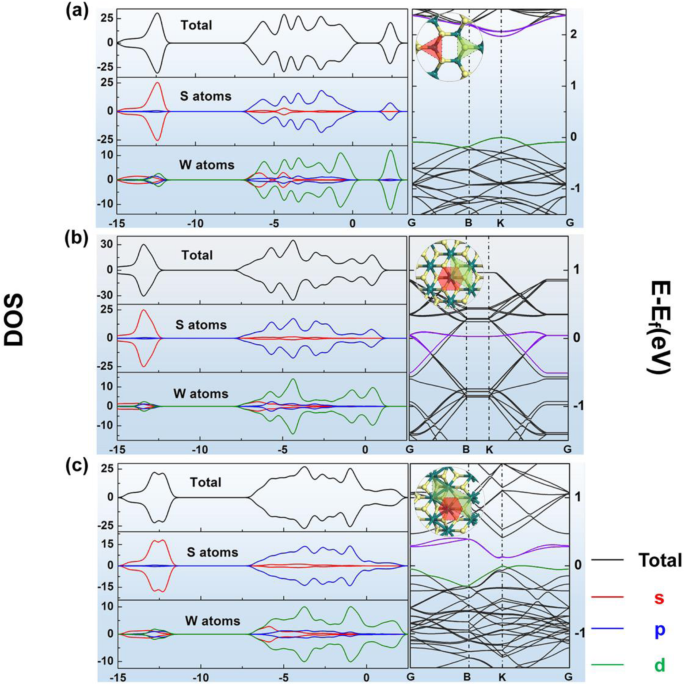
a的几何结构、DOS和能带结构结果 2H-WS2,b 1T-WS2 和 c 1T'-WS2;黄色球代表S,青色球代表W
c 上放置了一个氢分子 WS2平面上方的-轴建立氢吸附模型,并选择了几个几何对称性高的吸附位点。对于图 S2 (b) 和 (e) 中显示的 1T-WS2 的情况,有五个位点:刚好在上层 S 原子上方,刚好在下层 S 原子上方,刚好在 W 原子上方,在W原子和上层S原子,在W原子和下层S原子的键之上。对于1T'-WS2,这六种情况如图S2(c)和(f)所示。对于图S2(a)和(d)所示的2H-WS2,有四种情况:S原子位正上方、W原子位正上方、W原子和S原子位正上方、正上方六边形结构的中心。选择这些位点是因为它们是这些材料的高度对称位点。在给出几何优化和吸附能比较后,可以找到稳定的吸附位点。由于其相对较低的结构对称性,我们区分了吸附在 1T'-WS2 上的氢分子的姿势。氢分子被水平或垂直设置(如图 S3 所示),这使情况翻了一番。几何优化后,所有吸附能如表 S2 所示。根据吸附能结果选择最稳定的吸附位点。对于氢吸附过程,吸附能由以下函数计算:E 广告 =E 总 - E 垫 - E hyd,其中 E tot 是吸附氢分子的 WS2 的这三个相中的每一个的总能量,E mat(材料的能量)代表原始WS2的总能量,E hyd 代表孤立氢分子的总能量。根据这个关系,E 的绝对值更高 ad 导致吸附系统更加稳定。材料与目标气体分子之间的作用力也可以用E的绝对值来反映 广告。排斥力由 E 的正值表示 广告,而负值反映了吸引力。这种方法虽然不能得到准确的吸附能[34],但它可以反映氢与吸附材料相互作用的形式和强度。如上所述,每个氢分子在储氢应用中的理想吸附能是- 0.2至- 0.6 eV/H2在室温下[35]。
结果与讨论
对于所有这些材料的模型,在几何优化后可以找到能量最低的结构。单层 1T-WS2 和 2H-WS2 中所有 W-S 键的长度分别为 2.428 Å 和 2.402 Å。但是 1T'-WS2 中的那些是不等的,它们的长度大约为 2.453 Å、2.410 Å 和 2.490 Å。还可以发现优化的 1T' 模型中 W-W 键的长度约为 2.784 Å。优化的原始 WS2 的所有这三个相的能带结构如图 1 所示。对于金属 1T 相,没有带隙。对于1T'相,它具有半金属能带结构。而在 2H 相中,能带结构符合半导体的特性。这三个模型的部分态密度(PDOS)也如图 1 所示。 从 PDOS 结果可以看出,所有这三个图中 Sp 和 Wd 轨道的形状与总 DOS 的形状最相似,表明 Sp 和 Wd 轨道对总 DOS 有贡献,主要用于 WS2 的所有这三个阶段。 1T'-WS2 的 DOS 结果趋势与能带结构一致,与之前的研究一致 [32]。比较吸收的氢分子的不同位置,以在所有这三种模型中找到最稳定的。根据 E 选择位置 ad 和 Hirschfeld 对这三相结构中吸收的单个氢分子的列出情况的充电结果(E ad 和 Hirschfeld 电荷结果如图 2a-c 和表 S2 所示。对于1T WS2,它是站点3,对于1T'-WS2,它是站点1(如图2b,c所示),而对于2H-WS2,它是站点3(都显示在图2a和表中) S2-S3)。基于这些结果,首先,1T 相 WS2 不适合氢吸附,因为 E 1T WS2 上的氢的 ad 远比 0.6 eV 显着(表 S2)。这意味着很难从 1T WS2 表面释放吸附的氢分子。根据这一结果,以下研究不应关注这一阶段。 E 1T'相和2H相的ad结果约为- 0.27 eV,均在储氢应用的适用吸附能范围内。
<图片>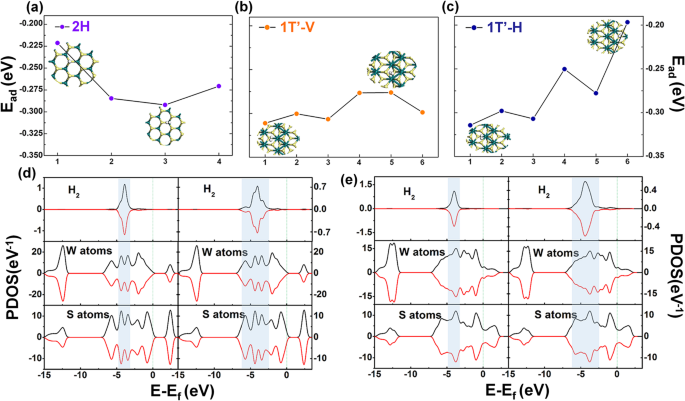
H2吸附系统对a的吸附能结果 2H-WS2 上的单个 H2,b 和 c 1T’-WS2 上的单个 H2; d 中 Ead 最低(左)或最高(右)情况的 PDOS 结果 2H 模型和 e 中的模型 1T’模型
为了进一步比较这两个阶段,进行了 PDOS 分析,如图 2d,e 所示。左侧部分显示了两相的最低 PDOS 吸附能,而右侧部分是最高的两个。最低或最高能量的两种情况都存在微小差异。在图 2d 的左侧,e(对应于最低吸附能),主峰都在 - 3 到 - 5 eV。而右侧部分(代表最高吸附能),它出现在- 2.5和- 6 eV之间。这种出现意味着氢分子的 PDOS 和 WS2 之间存在更大的叠加,这表明它们之间的相互作用更强。这些结果与吸附能结果非常吻合。然而,单氢分子情况下的PDOS结果仍不能很好地反映这两种材料吸氢性能的差异。
因此,我们对吸附在 1T' 和 2H-WS2 表面的不同数量的氢分子进行了研究。如图 S4 所示,我们在 1T' 和 2H-WS2 的表面设置了不同数量的氢分子(16、32、48 和 64)。对于1T'-WS2,当氢分子数在16以下时,每个氢分子都设置在最稳定的位置(1v位点)。考虑到多个 H2 分子之间潜在相互作用的影响,我们进一步讨论了当 2 或 3 个 H2 分子吸附 1T'-WS2 时 H2 的排列。对于两个氢分子,我们考虑了三种情况:相邻位点(2H2-1)、同侧分离位点(2H2-2)和异侧最近位点(2H2-3)。对于三个氢分子,有五种情况:同侧三个相邻位点(3H2-1);两个相邻,一个分开,都在同一侧(3H2-2);三个在同一侧分开(3H2-3);同一侧的两个邻居和另一侧的一个(3H2-4);两个在同一侧分开,一个在另一侧(3H2-5)。比较了每种情况下计算的吸附量(表 S4)。结果表明,在 1T'-WS2 的相邻位点上设置氢分子会使总吸附能大于分离情况。这意味着即使在同一吸附位点上随机设置H2分子也会带来不规则的吸附能变化。然而,当氢分子设置在 1T'-WS2 不同侧的最近位置时,没有明显的影响。基于这些结果,氢分子的设置遵循以下原则:当H2分子小于8时,氢分子设置在1T'-WS2两侧不相邻的吸附位点上;当数量为 8 到 16 时,无法避免相邻站点。仍然尽可能避免相邻的吸附位点。当氢分子在 17 到 32 之间时,其中 16 个设置在最稳定的位置(位置 1v),其余的设置在 W 原子的垂直上方(位置 3v)。当氢分子超过32个时,优先考虑这些氢分子之间的距离,避免形成氢分子团,如图S6所示。然后,水平或垂直放置将取决于单个氢的吸附能结果。因此,当 H2 在 33 和 48 之间时,前 16 个分子在 1v 位点,后 16 个分子在 3v 位点,其余在 4h 位点。当数字大于 48 时,前 16 个分子在 1v 位点,后 16 个分子在 3v 位点,第三个 16 个分子在 4h 位点,其余在 2h 位点。我们尽量将氢分子均匀地排列在这种结构的两侧,并确保每个氢分子之间的距离足够远。在2H相的条件下,类似于1T'-WS2的情况,当氢分子低于32时,每个氢分子都设置在上面讨论的最稳定的位置(位点3)。为避免氢分子间相互作用造成不一致的影响,当数量小于16时,氢分子被设置在不相邻的位置。但当数量在17到32之间时,应尽量避免相邻位置。当数量为在 33 和 64 之间,其余部分放置在六边形的中心(位置 4)。我们还尝试按照上述原则分配所有分子。另一方面,我们还考虑了具有高浓度 H2 分子的吸附系统的稳定性。当一个气体分子超过 16 个时,整个系统的稳定性也已通过分子动力学模拟进行了探索,如图 S7 所示。经过500步的分子动力学模拟,没有出现几何屈曲,总能量也几乎保持不变,整个系统具有很好的稳定性。
在给出几何优化后计算对氢分子的吸附能。如图 3 所示,无论 WS2 处于哪个相,当氢分子数增加时,总吸附能几乎呈线性增加。这意味着当氢分子数量增加时,材料与吸附分子之间的相互作用力不会发生太大变化。图 3a 中的绿色区域代表中等氢吸附能量区域。可以发现,2H-WS2 早于 1T' 相离开该区域。这意味着当吸附的 H2 分子的数量变得多余时,2H-WS2 将比 1T'-WS2 难以释放更多的氢分子,这意味着更小的氢容量。然后,如图3所示,在2H或1T'相的情况下,吸附氢分子的平均吸附能在- 0.2至- 0.6 eV范围内的氢分子数低于48或55 , 分别。这意味着氢在2H-WS2上的理论合理吸附量可达2.4 wt%,而在1T'相上可达2.7 wt%。这表明改变相可以有效提高 WS2 的储氢性能。两种WS2的平均吸附能在不超过8时先减小后增大。 容易理解,当材料吸附更多的气体分子时,气体分子与材料之间的平均相互作用力会变弱.然而,当氢分子数大于8时,平均吸附能增加的原因尚不清楚。
<图片>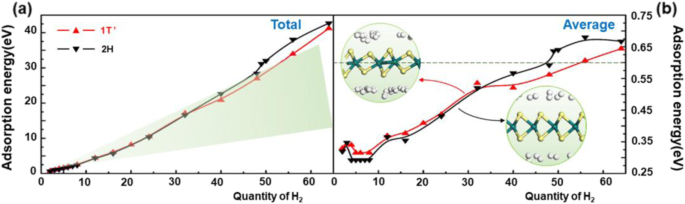
a 的图表 总吸附能和b 平均吸附能与吸附在 1T'- 和 2H-WS2 上的氢分子数量的函数关系
再次进行 PDOS 研究,如图 4 所示。可以发现,随着氢分子数的增加,吸附的氢分子的总 PDOS 会分散在 WS2 的两相中(尤其是当氢分子数为超过 16 个)。并且这些系统中吸附的单个氢分子的 PDOS 范围也变得更加广泛。但 W 原子和 S 原子的 PDOS 保持不变,这代表了这两种材料在吸附氢分子时的稳定性。结果还表明,随着氢分子数量的增加,氢分子与两个WS2分子之间的PDOS重叠面积增加。
<图片>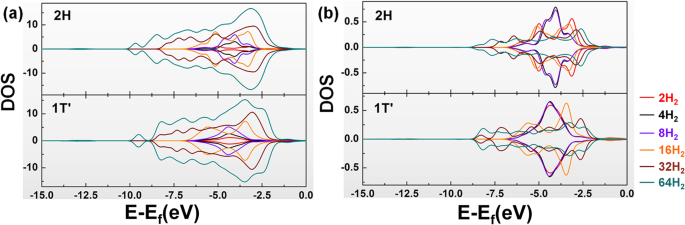
多个H2吸附系统对2H-WS2和1T'-WS2上a all 和b个单氢分子的PDOS结果
氢分子与WS2之间的相互作用变得更强。这就揭示了吸附氢分子数越多,平均吸附能越大的原因。
为了进一步探索氢分子与材料之间的相互作用,还进行了电子密度差(EDD)研究。如图 5(图 S5 中的平面图)所示,当 4、16、32 和 64 个氢分子吸附在 2H 或 1T'-WS2 上时,EDD 结果显示出来。橙色区域代表正值区域,表明有获得电子的趋势。而蓝色区域表示负区域,代表电子耗尽。对于 2H 和 1T'-WS2,橙色区域更可能出现在 S 原子附近,而蓝色区域更靠近 H 原子。当吸附 32 或 64 个氢时,趋势变得更加明显,如图 5c、d、g 和 h 所示。还可以观察到,当吸附更多的氢分子时,氢分子之间存在橙色和蓝色区域,表明吸附的 H2 分子之间存在相互作用。这增加了材料上每个氢分子的吸附力,增加了平均吸附能。此外,还有一点不容忽视,当更多的氢分子吸附在 1T'-WS2 上时,可以看到明显的蓝色区域(图 5 g,h)。而在2H情况下,这种现象并不明显。这表明 W 原子也经历了电子重新分配的过程。并且1T'-WS2 中的W 原子比2H-WS2 中的W 原子倾向于提供更多的电子来共享主要由氢分子提供的电子供应。基于此,作用在每个氢分子上的作用力都有一定程度的减弱。这可能是1T'-WS2在保证平均吸附力适度的情况下比2H-WS2可以容纳更多氢分子的原因。
<图片>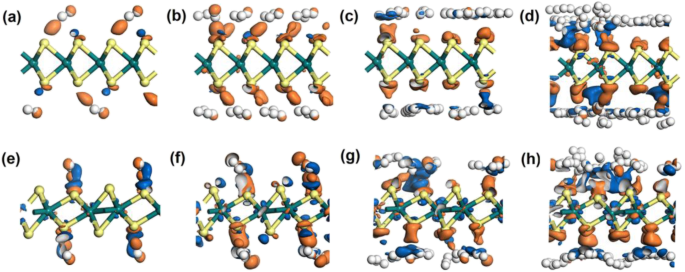
a的电子差分密度 2H-WS2 上的 4H2,b 2H-WS2 上的 16H2,c 2H-WS2 上的 32H2,d 2H-WS2 上的 64H2,e 1T′-WS2 上的 4H2,f 1T′-WS2 上的 16H2,g 1T'-WS2 上的 32H2 和 h 1T'-WS2 上的 64H2。等值面取值为 0.002 e/Å
结论
本文构建了2H、1T和1T'单层WS2的氢吸附模型。通过局部密度近似(LDA)探索它们对氢的吸附能力。然后,通过比较吸附多个氢分子时的吸附能,发现1T'-WS2可以比2H-WS2含有更多的氢分子,而平均吸附能在中等范围内(- 0.2至- 0.6 eV)。其合理的吸氢率可达2.7 wt%,高于2H-WS2的2.4 wt%,说明相对储氢的影响很明显,1T′相WS2的储氢容量大于2H对应。考虑到本研究中计算的所有结果,1T'相 WS2 是一种适用于氢吸附应用的材料。可为高集成储氢材料的研究提供理论参考。
数据和材料的可用性
所有数据完全可用,不受限制。
纳米材料