

长非编码 RNA MALAT1/microRNA-143/VEGFA 信号轴调节血管内皮损伤诱导的颅内动脉瘤
摘要
许多研究已经研究了一些长链非编码 RNA (lncRNA) 在颅内动脉瘤 (IA) 中的作用。本研究旨在阐明lncRNA转移相关肺腺癌转录本1(MALAT1)/microRNA-143(miR-143)/血管内皮生长因子-A(VEGFA)信号轴在血管内皮损伤诱导的IA中的机制。 .检测IA组织和正常动脉组织中MALAT1、miR-143和VEGFA的表达。检测组织中的基质金属蛋白酶9(MMP-9)、血清和组织中的血管性血友病因子(vWF)和血清中的内皮素-1(ET-1)。给模型化的 IA 大鼠注射沉默或过表达的 MALAT1 以检测血管内皮损伤。提取 IA 患者的血管内皮细胞并用沉默或过表达的 MALAT1 转染,以验证 MALAT1 对细胞活力和细胞凋亡的影响。 MALAT1、miR-143 和 VEGFA 之间的联系通过在线预测、荧光素酶活性和 RNA 下拉分析得到验证。在 IA 组织中发现 MALAT1 和 VEGFA 的过度表达以及 miR-143 的低表达。 MALAT1的下调抑制了IA大鼠的血压、ET-1、vWF和MMP-9的表达,以及血管内皮细胞的凋亡指数。下调的 MALAT1 抑制细胞凋亡并促进 IA 中血管内皮细胞的活力。 MALAT1 与 miR-143 结合,miR-143 靶向 VEGFA。本研究提示MALAT1通过与miR-143竞争性结合上调VEGFA表达,从而促进IA中血管内皮细胞的凋亡并减弱其活力。
介绍
颅内动脉瘤(Intracranial aneurysm,IA)又称脑动脉瘤,是由于脑动脉腔或动脉壁局部异常增宽引起颅内压升高所致[1]。 IA是一种死亡率和发病率高的严重疾病,在一般人群中的患病率约为1%~3%[2]。 IA 的主要临床特征是脑血管痉挛、自发性脑出血和动眼神经麻痹 [3]。目前,导致IA发生发展的常见危险因素包括血流动力学障碍、基因、衰老、感染和先天性因素[4]。主要的临床治疗方法,主要是手术夹闭和/或血管内弹簧圈栓塞,用于预防动脉瘤破裂 [5]。然而,IA的具体机制仍有待阐明,反映急需更有效的IA管理方法。
长链非编码 RNA (lncRNA) 长度超过 200 个核苷酸,属于一类非编码 RNA [6]。据报道,lncRNA 生长停滞相关的 lncRNA 1 在 IA 中的表达下调 ancung 腺癌转录本 1 (MALAT1) 是一种高度富集和广泛表达的 lncRNA,其长度约为 8000 nt [7]。 MALAT1 已被证明可调节胸主动脉瘤中的平滑肌功能障碍 [8]。此外,一项研究提出了 IA 中 lncRNA 和信使 RNA (mRNA) 的异常表达,lncRNA-mRNA 共表达网络为寻找 IA 的发病机制提供了线索 [9]。 MALAT1 已被建议通过靶向人骨髓间充质干细胞中的 miRNA-143 (miR-143) 表达来促进 osterix 表达并调节成骨分化 [10]。一项研究还提出了 miR-143/145 簇在逆转平滑肌细胞中 krüppel 样因子 5 的调节及其在 IA 中的收缩性和增殖中的作用 [11]。根据 Feng 等人的研究,循环过程中 miR-143/145 的下调和较高的基质金属蛋白酶 9 (MMP-9) 水平可能与 IA 的形成和破裂有关 [12]。一项分析表明,最不受控制的 miRNA(miR-143 和 miR-145)常见于作为信号通路的目标基因,例如血管内皮生长因子 (VEGF) 和其他调节细胞运动或粘附的基因 [13] .一项研究揭示了血管内皮生长因子-A (VEGFA) 变异在 IA 中的预测重要性 [14]。因此,我们试图评估MALAT1/miR-143/VEGFA信号轴在血管内皮损伤诱导的IA中的作用机制。
材料和方法
道德声明
该研究得到了中国科学技术大学生命科学与医学部中国科学技术大学附属第一医院机构审查委员会的认可,并遵循了赫尔辛基宣言的原则。参与者在本研究中提供了书面知情同意书。所有动物实验均符合美国国立卫生研究院实验动物护理和使用指南。该方案经中国科学技术大学第一附属医院生命科学与医学部动物实验伦理委员会批准。
学习科目
选取中国科学技术大学生命科学与医学部中国科学技术大学第一附属医院经影像学检查确诊并接受神经外科夹闭的20例IA患者(IA组)进行实验。男11例,女9例,年龄43.27±6.25 。对 IA 组织取样。同时切除20例杏仁核、海马硬化所致颞叶癫痫患者(对照组)颞极颞叶皮质血管组织。切除的组织经术后病理组织学检查为正常动脉组织,男13例,女7例,年龄44.18±5.91 。 IA组和对照组之间在性别和年龄方面没有显着差异(均P> 0.05)。所有受试者均于术前早晨同一时间空腹采集静脉血(2管)。
建立IA大鼠模型
60 只清洁级 Sprague-Dawley 雄性大鼠(湖南 SJA 实验动物有限公司,湖南,中国),体重在 200 至 250 g 之间,饲养 7 天(25±2 °C,相对湿度为 65-70 %,12 h 的明暗循环,自由饮水和食物摄入)。用猪胰弹性蛋白酶将大鼠滴入颈外动脉和分叉动脉壁周围。颈外动脉在颈外动脉分支约1.5 mm处用两条手术线结扎。在两条线之间切断颈外动脉,在颈外动脉盲段形成颈内动脉瘤。手术后给大鼠喂食1%生理盐水1周。 1 个月后行脑血管造影,观察动脉瘤形成。
IA大鼠模型建立后,50只大鼠随机分为空白组(n =10,模型大鼠立体定向注射100 μL磷酸盐缓冲盐水(PBS),每天一次),短发夹RNA(sh)-阴性对照(NC)组(n =10,模型大鼠立体定向注射100 μL sh-MALAT1 NC,每天一次),sh-MALAT1组(n =10,模型大鼠立体定向注射100 μL sh-MALAT1质粒,每天一次),过表达(Oe)-NC组(n =10,模型大鼠立体定向注射100 μL Oe-MALAT1 NC质粒,每天一次),Oe-MALAT1组(n =10,模型大鼠每天一次立体定向注射 100 μL Oe-MALAT1 质粒)[15]。以上质粒由上海基因化学有限公司(中国上海)合成。
大鼠血压测试
分别于术后第1、4、12周测量大鼠尾动脉血压。测量血压前,将大鼠置于恒温加热装置中片刻,以防止外界温度的干扰。其次,将大鼠置于特制的鼠笼中静默几分钟,防止活动受到干扰。如果血压变化幅度较大,则在不同时间测定2-3次取平均值。
动脉瘤组织获取
3 个月后,用1%戊巴比妥钠(40 mg/kg)腹腔注射麻醉大鼠,取静脉血。对大鼠实施安乐死,开胸,将左心室插管至主动脉,切开腔静脉放血。同时用30 mL含肝素钠的生理盐水快速心脏灌注冲洗血液,然后将10 mL 10%聚甲醛/0.1 M PBS(pH 7.4)注入脑内。灌注固定后,打开大鼠脑。仔细观察颅底动脉循环,分离动脉瘤组织,显微镜下观察动脉瘤的变化。
酶联免疫吸附测定 (ELISA)
采用ELISA试剂盒检测血清相关指标。将采集的血样置于37 ℃恒温箱中1 h,3000 r/min离心10 min。内皮素-1(ET-1)和血管性血友病因子(vWF)表达的检测按照试剂盒说明书进行(所有试剂盒均购自江苏南京建诚生物工程研究所)。
苏木精-伊红 (HE) 染色
样品用10%福尔马林固定24 h以上,石蜡块保存。石蜡块用二甲苯脱蜡20 min,用梯度递减系列酒精(100%、95%、80%、75%)脱水l min,苏木精染色10 min。然后用蒸馏水冲洗组织,用盐酸乙醇分化30 s,在50 ℃的温水中浸泡5 min。用伊红溶液染色,用蒸馏水冲洗,用70%和90%酒精脱水,用二甲苯澄清,用中性树胶密封。高倍显微镜下观察组织形态。
透射电子显微镜观察
备用组织用2.5%戊二醛和1%锇酸固定,脱水后用Epon812树脂包埋。半薄切片用甲苯蓝染色,修整,制成超薄切片。切片用醋酸双氧铀和柠檬酸铅染色,JME-2000EX透射电子显微镜(日立高新技术有限公司,上海,中国)观察。
末端脱氧核苷酸转移酶介导的脱氧尿苷三磷酸-生物素缺口末端标记 (TUNEL) 染色
在TUNEL试剂盒(Roche,Basel,Switzerland)的基础上,暗示TUNEL染色观察IA大鼠的内皮细胞凋亡。制备好的大鼠动脉瘤切片用二甲苯洗涤2次(5 min/次),并用递减系列酒精(100%、95%、80%、75%)脱水3次(5 min/次)。组织用不含 DNase 的 20 μg/mL 蛋白酶 K 溶液处理 15-30 min,滴加 50 μL TUNEL 反应溶液 60 min,然后用 50 μL 二氨基联苯胺 (DAB) 在 25 °C 显色 10 min。然后,切片用苏木精复染,梯度酒精脱水,二甲苯澄清,中性树胶密封。光学显微镜下观察切片并计算细胞凋亡指数。
动脉瘤血管内皮细胞的分离鉴定
根据 Boscolo 等人进行的方法进行内皮细胞分离。 [16]。 IA组织被切成3 mm 2 碎片。将组织与 0.1% 胶原酶 B/0.1% 分散酶 (Roche) 在 37 °C 下孵育 25 分钟。然后,将预分离的组织用 2 mL 移液器研磨 2 分钟,并用 100-μm 过滤器(Thermo Fisher Scientific, Rockford, IL, USA)过滤。随后,将细胞悬液离心,然后重悬于 MV2 培养基(包括生长因子和 20% 胎牛血清)(PromoCell,Heidelberg,Germany)中。细胞接种于 1 × 10 4 1 μg/cm 2 包被的培养瓶中的细胞/mL 纤连蛋白。按照 Jackson 等人描述的方法。 [17],80-100% 汇合的细胞用由 Ulex europaeus Agglutinin I (UEA) (Vector Laboratories, Ltd., Peterborough, UK) 包被的珠子 (Dynabeads M-450 Tosylactived, Oxoid, Hampshire, UK) 分离.粘附在凝集素包被珠上的内皮细胞用磁性颗粒浓缩器聚集,未结合的细胞用基础培养基洗涤。 UEA阳性细胞重悬于培养基中,接种于纤连蛋白包被的培养瓶中,以提高细胞的粘附和生长速度。
细胞在 MV2 中在纤连蛋白包被的室载玻片上生长。当细胞汇合度达到 80-100% 时,将细胞固定在 4 °C 的丙酮中,并用 1% Triton X-100 处理 5 分钟,然后用 0.5% 牛血清白蛋白 (BSA) 处理 15 分钟。将细胞滴入针对 vWF 的一抗(1:300,Abcam,Cambridge,MA,USA)并孵育 2 小时(在没有一抗的情况下进行 NC),滴入辣根过氧化物酶推测的免疫球蛋白 G(1: 150,Abcam)并孵育 30 分钟。细胞用50 μL DAB在25 °C显色5 min,苏木精复染,0.1%盐酸分化,酒精脱水,二甲苯清除,中性树胶封闭。细胞干燥后倒置显微镜下拍照。
细胞分组和转染
将处于对数期的动脉瘤血管内皮细胞分为5组:空白组(未经任何处理的动脉瘤血管内皮细胞)、sh-NC组(sh-MALAT1 NC质粒转染的动脉瘤血管内皮细胞)、sh-MALAT1组( sh-MALAT1质粒转染动脉瘤血管内皮细胞)、Oe-NC组(Oe-MALAT1 NC质粒转染动脉瘤血管内皮细胞)、Oe-MALAT1组(Oe-MALAT1质粒转染动脉瘤血管内皮细胞)。上述质粒由Genechem合成。按照lipofectamine TM 说明书进行细胞转染 2000 试剂(11668-027,Invitrogen,Carlsbad,California,USA)。
3-(4, 5-Dimethylthiazol-2-Yl)-2, 5-Diphenyltetrazolium Bromide (MTT) 检测
将各组血管内皮细胞以3×10 4 的密度接种于96孔板上 细胞/mL,并在 37 °C、5% CO2 条件下培养 48 小时。每组设5个平行孔,每孔加入20 μL新鲜MTT溶液(5 mg/mL,Sigma, St. Louis, MO, USA)。反应4小时后,将细胞与200 μL二甲亚砜混合。完全溶解后,用酶标仪(BioRad,Hercules,California,USA)在490 nm处测定各组细胞的光密度值。
流式细胞术
通过碘化丙啶 (PI) 染色测试细胞周期分布。血管内皮细胞被分离、离心、用预冷的 75% 乙醇重悬,并在 - 20 °C 下固定过夜。离心细胞以丢弃上清液。将细胞加入450 μL PBS中,加入100 μL Rnase A,400 μL PI在4 °C避光染色30 分钟。流式细胞仪(FACSCalibur, Becton, Dickinson and Company, NJ, USA)用于检测细胞周期分布。
通过Annexin V/PI双染色检测细胞凋亡。收集分离的内皮细胞并用PBS洗涤3次。细胞用 100 μL 预冷的 1×结合缓冲液重悬,并分别与 5 μL Annexin 和 5 μL PI 混合。通过流式细胞仪检测细胞凋亡。以 AnnexinV 为横轴,PI 为纵轴,左上象限代表机械损伤细胞 (AnnexinV − /PI + ),晚期凋亡细胞或坏死细胞的右上象限 (AnnexinV + /PI + ),阴性正常细胞的左下象限 (AnnexinV − /PI − ),以及早期凋亡细胞的右下象限 (AnnexinV + /PI − )。总凋亡细胞(AnnexinV + /PI − 和 AnnexinV + /PI + ) 计算并以百分比表示。
逆转录定量聚合酶链反应 (RT-qPCR)
组织和细胞中的总 RNA 由 Trizol(宝生物技术有限公司,大连,中国)提取,并测定 RNA 的浓度和纯度。 RNA逆转录为互补DNA的过程按照逆转录试剂盒(K1621,Fermentas,Maryland,NY,USA)的说明进行。 MALAT1、miR-143 和 VEGFA 引物序列(表 1)由 Genechem 设计和组成。为了评估 lncRNA、miRNA 或 mRNA 的表达,使用 SYBR GreenPCR Master Mix(Takara,Tokyo,Japan)和 Roche 实时 PCR 系统(Roche)进行 RT-qPCR。 U6 被设置为 miR-143 的内部参数,而 MALAT1 和 VEGFA 以甘油醛-3-磷酸脱氢酶 (GAPDH) 为内部参数。目的基因的相对转录水平由2 −△△Ct 计算 方法。
Western Blot 分析
从组织和细胞中提取总蛋白质。蛋白质浓度按照二辛可宁酸试剂盒(湖北武汉博斯特生物科技有限公司)说明书测定。通过使用 10% 聚丙烯酰胺凝胶 (Boster Biological Technology) 的电泳分离蛋白质。将膜转移到聚偏二氟乙烯膜上,然后用 5% BSA 密封 1 小时。将膜与针对 Ki-67 (1:1000)、VEGFA (1:1000)、vWF (1:1000) 和基质金属蛋白酶 (MMP)-9(1:1000,Abcam,Cambridge,UK)的一抗一起孵育, Cyclin D1 (1:1000), Bax (1:1000), and Bcl-2 (1:1000, Santa Cruz Biotechnology, Santa Cruz, California, USA), and GAPDH (1:2000, Jackson Immuno Research, Grove,宾夕法尼亚州,美国)和二抗(1:500,Jackson Immuno Research)用辣根过氧化物标记。膜由奥德赛二色红外荧光扫描成像系统获得,条带灰度值由Quantity One图像分析软件测量。
双荧光素酶报告基因检测
MALAT1和miR-143的结合位点由生物信息学网站(https://cm.jefferson.edu/rna22/Precomputed/)预测和阐述。 MALAT1和miR-143之间的结合关系通过荧光素酶报告基因检测进一步验证。将合成的 MALAT1 3'非翻译区 (3'UTR) 基因片段引入 pmiR-Report 荧光素酶报告载体 (Thermo Fisher Scientific),通过内切酶位点 Bamh1 和 Ecor1 生成 MALAT1 野生型 (MALAT1-WT)。在 MALAT1-WT 上设计了序列的互补序列突变位点,并将目标片段插入 pmiR-Report 荧光素酶报告载体中,在限制性内切酶消化后通过 T4 DNA 连接酶产生 MALAT1 突变型(MALAT1-MUT)。正确测序的 MALAT1-WT 和 MALAT1-MUT 与模拟 NC 和 miR-143 模拟共转染到血管内皮细胞中。转染后48 h收获细胞并裂解,通过荧光素酶检测试剂盒(BioVision,San Francisco,CA,USA)和光度计(Glomax20/20,Promega,Madison,Wisconsin,USA)检测荧光素酶活性。
通过生物信息学网站(http://www.targetscan.org/vert_72/)预测miR-143和VEGFA的靶标关系以及miR-143和VEGFA 3'UTR的结合位点。将含有 miR-143 结合位点的 VEGFA 3'UTR 启动子区序列复合并克隆到 pmiR-Report 荧光素酶报告载体中以产生 VEGFA-WT。在此报告基因的基础上,VEGFA-WT 和 miR-143 的结合位点发生突变,形成 VEGFA-MUT。 VEGFA-WT 或 VEGFA-MUT 报告基因与模拟 NC 或 miR-143 模拟混合,然后共转染血管内皮细胞 48 h。然后裂解细胞,用荧光素酶检测试剂盒检测荧光素酶活性。
RNA 下拉分析
为了验证 miR-143 和 MALAT1 之间的结合关系,进行了 RNA-pull down 试验。设计了三个生物素标记的 miRNA 序列 Bio-miR-143-WT、Bio-miR-143-MUT 和 Bio-miR-NC,并委托给 GenePharma 公司(中国上海)。将这些生物素化的寡核苷酸转染到细胞中 48 h。然后,收获细胞并与特定细胞裂解物(Ambion,Austin,Texas,USA)一起孵育 10 分钟。之后,将裂解液与预包被无 RNase 和酵母 tRNA(均来自 Sigma)的 M-280 链霉亲和素珠在 4 °C 下孵化 3 小时,然后用冷裂解液清洗两次,用低浓度清洗 3 次。盐缓冲液,一次用高盐缓冲液。拮抗 miR-143 探针被建立为 NC。 Trizol提取总RNA,RT-qPCR检测MALAT1富集水平。
统计分析
所有数据均由 SPSS 21.0 软件(IBM Corp. Armonk, NY, USA)阐述。测量数据以平均值±标准差表示。两组比较采用独立样本t 检验,而多组间的比较采用单向方差分析(ANOVA),成对比较采用 Tukey 多重比较检验。通过卡方检验确定MALAT1表达与IA临床病理特征之间的关系。 P 值小于 0.05 表示差异有统计学意义。
结果
在 IA 组织中 MALAT1 和 VEGFA 过表达,而 miR-143 下调
ELISA检测IA组和对照组血清中ET-1和vWF的表达,结果表明IA组与对照组相比ET-1和vWF表达升高(均P <0.05) (图 1a, b).
<图片>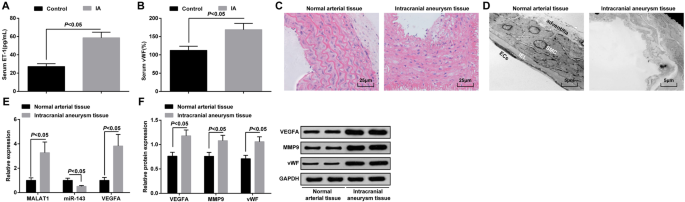
MALAT1 和 VEGFA 过表达,而 miR-143 在 IA 组织中下调。 一 ELISA检测IA患者和颞叶癫痫患者血清中ET-1的表达。 b ELISA检测IA患者和颞叶癫痫患者血清中vWF的表达。 c HE染色对IA组织和正常动脉组织的病理观察。 d 通过透射电子显微镜对IA组织和正常动脉组织的形态学观察。 e 通过 RT-qPCR 检测 IA 组织和正常动脉组织中 MALAT1、miR-143 和 VEGFA mRNA 的表达。 f 通过蛋白质印迹分析在 IA 组织和正常动脉组织中的 VEGFA、MMP-9 和 vWF 蛋白表达。内皮细胞 (EC)、内部弹性层 (IEL)、平滑肌细胞 (SMC)。测量数据表示为平均值±标准偏差;组间比较采用独立样本t 测试
HE染色观察IA组织病理变化。正常动脉组织内胚层、内皮细胞、平滑肌细胞未见明显损伤,细胞排列整齐,结构完整。 IA组织内皮细胞受损,平滑肌细胞退化,动脉壁变细,弹力纤维破裂,炎症细胞浸润(图1c)。
透射电镜观察IA和正常动脉组织的超微结构变化。发现内皮细胞完整,外膜结构完整;正常动脉组织未见内弹力膜破裂或平滑肌细胞凋亡。在IA组织中,表现为血管壁严重变性,主要表现为大部分内皮细胞消失,内弹力层严重破坏,平滑肌细胞严重受损退化,血管外膜功能紊乱。 (图1d)。
RT-qPCR 用于确定 MALAT1、miR-143 和 VEGFA mRNA 的表达,而蛋白质印迹分析用于 IA 组织和正常动脉组织中的 VEGFA、MMP-9 和 vWF 蛋白表达。结果表明,与正常动脉组织相比,IA 组织中 MALAT1、VEGFA、MMP-9 和 vWF 表达水平升高,miR-143 表达降低(所有 P <0.05) (图 1e, f).
Hunt-Hess 等级、内皮损伤程度和吸烟史与 IA 组织中的 MALAT1 表达相关
根据MALAT1的中位表达,将患者分为两组:低表达组和高表达组。分析MALAT1表达与IA患者临床病理特征的关系。结果提示Hunt-Hess分级、内皮损伤程度、吸烟史与MALAT1表达相关(均P <0.05),而年龄、性别和手术方式与 MALAT1 表达无关(所有 P> 0.05)(表 2)。
下调 MALAT1 抑制血压、ET-1、vWF 和 MMP-9 的表达,以及大鼠血管内皮细胞的凋亡指数与 IA
如表 3 所示,MALAT1 敲低降低,而 MALAT1 恢复在第 4 周和第 12 周升高血压。
ELISA检测显示IA大鼠血清中MALAT1下调降低,MALAT1上调升高ET-1和vWF表达(图2a、b)。
<图片>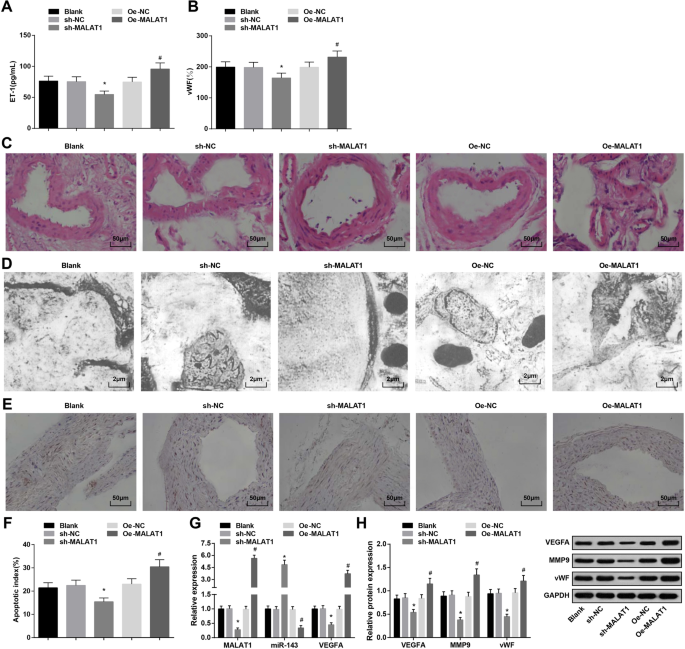
下调的 MALAT1 抑制血压、ET-1、vWF 和 MMP-9 的表达,以及 IA 大鼠血管内皮细胞的凋亡指数。 一 ELISA法检测大鼠血清中ET-1的表达。 b ELISA法检测大鼠血清中vWF的表达。 c HE染色观察大鼠IA组织病理变化。 d 透射电子显微镜观察大鼠 IA 组织的超微结构。 e 通过 TUNEL 染色观察血管内皮细胞的凋亡。 f 大鼠血管内皮细胞凋亡指数。 g RT-qPCR 检测大鼠 IA 组织中 MALAT1、miR-143 和 VEGFA mRNA 的表达。 h 通过蛋白质印迹分析大鼠IA组织中VEGFA、MMP-9和vWF蛋白的表达。 * P <0.05 vs. sh-NC 组,# P <0.05 与 Oe-NC 组相比。测量数据以平均值±标准差表示,多组间比较采用单因素方差分析和Tukey多重比较检验
HE染色观察各组IA组织病理变化。空白组、sh-NC组和Oe-NC组内膜损伤、内皮细胞脱落、平滑肌细胞退化、细胞和层数减少、动脉壁变薄。 sh-MALAT1组颅内动脉内皮细胞、内皮细胞、平滑肌细胞层、外膜层损伤轻微但排列整齐。 Oe-MALAT1组内膜层消失,内皮细胞脱落,弹力纤维断裂,炎症细胞浸润(图2c)。
透射电镜观察各组大鼠IA组织。结果显示空白组、sh-NC组和Oe-NC组内皮细胞变性、内皮下层解体、内弹性层消失、平滑肌细胞减少。 sh-MALAT1 组表现出扁平内皮细胞、椭圆形细胞核和增加的胶原纤维,但没有弹性层。 Oe-MALAT1组内皮细胞消失,弹性层与肌层分离,解体进入管腔(图2d)。
TUNEL染色检测IA大鼠血管内皮细胞凋亡指数。沉默MALAT1降低了血管内皮细胞的凋亡指数,而过表达的MALAT1则表现出相反的作用(图2e,f)。
RT-qPCR 检测 MALAT1、miR-143 和 VEGFA mRNA 表达,以及 IA 组织中 VEGFA、MMP-9 和 vWF 蛋白表达的蛋白质印迹分析表明 MALAT1 消除抑制 MALAT1、VEGFA、MMP-9 和 vWF 表达,并提高了 miR-143 的表达。相反,MALAT1 升高对这些基因表达产生相反的影响(图 2g,h)。
MALAT1 的低表达促进 IA 中血管内皮细胞的活力并抑制其凋亡
采用免疫组织化学染色检测内皮特异性标志物 vWF 的表达。表现为IA血管内皮细胞胞浆覆盖有棕色细小颗粒,为阳性,而其NC组胞浆无棕色颗粒。结果证实培养的细胞为内皮细胞(图3a、b)。

Low expression of MALAT1 advances viability and restrains apoptosis of vascular endothelial cells in IA. 一 vWF immunohistochemical staining in IA vascular endothelial cells:IA vascular endothelial cells were covered with fine yellow particles. b vWF immunohistochemical staining in IA vascular endothelial cells:IA vascular endothelial cells showed no brown particles in the NC group. c Vascular endothelial cell viability in each group by MTT assay. d Protein expression of CyclinD1 and Ki-67 in each group by western blot analysis. e Cell cycle changes in each group by PI staining. f Cell apoptosis rate in each group by Annexin V/PI double staining. g Bax and Bcl-2 protein expression in each group by western blot analysis. * P <0.05 vs. the sh-NC group, # P <0.05 vs. the Oe-NC group. Measurement data were depicted as mean ± standard deviation, and comparisons among multiple groups were assessed by one-way analysis of variance followed with Tukey’s multiple comparisons test
MTT assay, flow cytometry, together with western blot analysis were utilized to test vascular endothelial cell viability and apoptosis. It was displayed that MALAT1 diminution promoted vascular endothelial cell viability (heightened Cyclin D1 and Ki-67 expression) and depressed apoptosis (decreased G0/G1 phase cells and increased S and G2/M phase cells, reduced Bax and elevated Bcl-2 expression). However, MALAT1 upregulation functioned in an opposite way to that of MALAT1 diminution on cell viability and apoptosis (Fig. 3c–g).
MiR-143 Is Bound to MALAT1 and VEGFA Is a Target Gene of miR-143
MALA1, miR-143, and VEGFA expression in vascular endothelial cells of each group were verified through RT-qPCR and western blot analysis. The results presented that MALA1 knockdown depressed MALA1 and VEGFA expression and enhanced miR-143 expression. On the contrary, MALA1 upregulation led to the increment in MALAT1 and VEGFA expression and the reduction in miR-143 expression (Fig. 4a, b).

MiR-143 is bound to MALAT1 and VEGFA is a target gene of miR-143. 一 MALAT1, miR-143, and VEGFA mRNA expression in vascular endothelial cells of aneurysm in each group. b VEGFA protein expression in vascular endothelial cells of aneurysm in each group. c The binding sites of MALAT1 and miR-143 predicted by the bioinformatics website. d The regulatory relation of MALLA1 and miR-143 validated by dual luciferase reporter gene assay. e The binding relationship between MALAT1 and miR-143 verified by RNA-pull down assay. f The binding sites of miR-143 and VEGFA predicted by the bioinformatics website. g The regulatory relation of miR-143 and VEGFA validated by dual luciferase reporter gene assay. * P <0.05 vs. the sh-NC group, # P <0.05 vs. the Oe-NC group. Measurement data were depicted as mean ± standard deviation, comparisons between two groups were assessed by independent sample t test, and comparisons among multiple groups were assessed by one-way analysis of variance followed with Tukey’s multiple comparisons test
The specific binding region between MALAT1 and miR-143 was divined by online analysis software (Fig. 4c). The results of dual luciferase reporter gene assay revealed that the luciferase activity was impaired in the MALAT1-WT + miR-143 mimic group versus the MALAT1-WT + mimic-NC group (P <0.05). However, there was no distinct difference in luciferase activity in the MALAT1-MUT + miR-143 mimic group relative to that in the MALAT1-MUT + mimic-NC group (P> 0.05), indicating that miR-143 was specifically bound to MALAT1 (Fig. 4d). The results of RNA-pull down assay reported that the enrichment level of MALAT1 in the Bio-miR-143-WT group was heightened compared to the Bio-miR-NC group (P <0.05), but there was no distinct difference in MALAT1 enrichment level in the Bio-miR-143-MUT group (P> 0.05) (Fig. 4e).
Bioinformatics software divined a targeted relationship between miR-143 and VEGFA (Fig. 4f). The results of luciferase activity showed that the relative luciferase activity repressed after VEGFA-WT and miR-143 mimic co-transfected into vascular endothelial cells (P <0.05). However, the relative luciferase activity of vascular endothelial cells was not affected by co-transfection of VEGFA-MUT and miR-143-mimic (P> 0.05) (Fig. 4g). It was indicated that VEGFA was a direct target gene of miR-143.
Discussion
IA is a serious intracranial disease, which mainly leads to subarachnoid hemorrhage [18]. A previous study has demonstrated the involvements of lncRNA-related competitive endogenous RNA networks in IA [19]. Also, a recent study has provided a proof that functional polymorphism in miR-143/145 gene promoter region is connected with the risk of IA [20]. In a study conducted by Xu et al. , it is shown that overexpression of angiogenic factors, such as VEGFA, may be related to IA formation and rupture [21]. In order to explain the molecular mechanism of MALAT1 in IA, a series of assays have been conducted and the results revealed that IA induced by vascular endothelial injury was regulated by MALAT1/miR-143/VEGFA signal axis.
Firstly, our study has provided substantial evidence that MALAT1 and VEGFA are upregulated and miR-143 is downregulated in IA tissues. A recent study has presented that MALAT1 is one of the most upregulated lncRNAs in the process of cerebral ischemia [22]. Another study has presented that MALAT1 is upregulated in ovarian cancer cells and intends to participate in the processes of ovarian cancer cell apoptosis, migration, and proliferation [23]. A study concerning to the expression profile of unruptured and ruptured IA has demonstrated that the expression of angiogenic factors such as VEGFA is upregulated in ruptured aneurysm [21]. Moreover, a clinical study has presented that the miR-143/145 cluster of IA patients is downregulated compared to healthy subjects [11]. In addition, it is previously discussed that miR-143/145 takes part in various biological processes associated with aneurysm formation and is downregulated in patients with IA [20]. All these findings are more or less echoed with the previous exploration outcomes.
Except for the abovementioned findings, this study has also explored the functional role of MALAT1 in IA through gain-off-function and loss-of-function assays. It could be summarized that downregulation of MALAT1 reduced blood pressure, serum levels of ET-1, and vWF and MMP-9 expression in IA tissues. It has been suggested previously that downregulation of MALAT1 restrains the upregulation of the glucose-induced ET-1 transcription product [24]. Also, it is reported that ectopic MALAT1 expression is the inducer of vWF generation [25]. Another study has verified that the depletion of MALAT1 in bone marrow-derived macrophages inhibits the expression of MMP-9 [26].
Also, cell experiments have been conducted for further confirmation of the functions of MALAT1 in IA. The results have suggested that MALAT1 knockdown promoted vascular endothelial cell viability and depressed apoptosis in IA. Similarly, it has been documented that disturbance of MALAT1 improves aortic mural architecture and retards aneurysm growth [8]. Supplementary to our study finding, there is research highlighting that poor expression of MALAT1 induces apoptosis and restrains proliferation of acute myeloid leukemia cells [27]. Another study has also demonstrated the inhibitory effects of MALAT1 knockdown on proliferation of human osteoarthritis cartilage cells [28]. Besides that, a prior research generally confirms that downregulation of MALAT1 can induce apoptosis and attenuate the proliferation of glioma cells [29]. Moreover, low expression of MALAT1 induced by RNA interference promotes apoptosis and suppresses proliferation of multiple myeloma cells [30]. Collectively, these studies have explained the molecular mechanism of MALAT1 in IA to some extent.
In addition, this study has evidenced that miR-143 is bound to MALAT1 and VEGFA is a target gene of miR-143. Similarly, a paper contends that MALAT1 binds directly to miR-143 and suppresses its expression [10].朱等人。 have found that MALAT1 exerts its roles via interacting with miR-143 in cervical carcinoma cells [31]. It is further confirmed that MALAT1 indirectly modulate VEGFA via miR-200b-3p [32]. Moreover, another study has suggested that miR-143-3p mediates ZFPM2 effect on a number of protein targets in blood, including VEGFA [33]. Nevertheless, the interactions among MALAT1, miR-143, and VEGFA in IA have not been discussed and need further study.
Conclusion
From these results, it is clear that MALAT1 knockdown depresses apoptosis and promotes viability of vascular endothelial cells in IA by modulating miR-143/VEGFA axis. The co-expression network suggests the connection between MALAT1 and miR-143 with the involvement of VEGFA. The findings in this study partially disclose the pathogenesis of IA initiation and progression, and the studied targets may be a notably potential entry point to reveal pathology of IA from another perspective. Limitedly, further large-scale studies are required to comprehensively illustrate the mechanisms of MALAT1/miR-143/VEGFA axis in IA.
纳米材料