

小分子在五石墨烯上吸附行为的第一性原理研究
摘要
使用第一性原理计算研究了小分子 CO、H2O、H2S、NH3、SO2 和 NO 在原始五石墨烯 (PG) 上的气体吸附行为,以探索它们作为先进气敏材料的潜力。结果表明,除CO外,H2O、H2S、NH3和SO2物理吸附在五石墨烯表面,具有相当大的吸附能和适度的电荷转移,而NO容易化学吸附在五石墨烯表面.此外,PG在吸附H2O、H2S、NH3、SO2和NO后可以有效改变其电子性质,五石墨烯具有通过电荷转移机制应用于气体传感器的潜力。
背景
气体传感,尤其是污染或有毒气体传感,一直是与环境污染监测、工业控制、农业生产和医学诊断等领域应用相关的研究热点[1]。最近,越来越多的二维材料被预测和合成 [2,3,4]。二维材料已被广泛研究并用作气敏元件,因为它们具有迷人的特性,例如大表面积、超高载流子迁移率和低电噪声 [5]。据报道,二维材料吸附某些特定的气体分子后,其电子性质可能会发生变化[6, 7]。
在二维材料中,特别是石墨烯及其类似物因其卓越的物理特性和在纳米电子学和纳米力学中的应用潜力而备受关注 [8,9,10,11,12]。尽管石墨烯被广泛认为是最适合下一代电子器件的主体材料之一 [13],但稳定的 sp 2 碳键的杂化和零间隙特性使其气体吸附效率低下,这不利于气体传感器的设计。此外,石墨烯是一种具有优异导电性的导体[8]。与半导体相比,在气体吸附过程中电阻信息难以测量,石墨烯对气体浓度变化不敏感。因此,石墨烯需要被功能化以打开带隙并充当半导体 [10]。由于实验表征的局限性,人们通过第一性原理计算广泛研究了分子在石墨烯表面的吸附行为,这对石墨烯的应用具有重要意义[14,15,16]。
五石墨烯 (PG) 可以从大块 T12 碳中剥离,是最近提出的石墨烯同素异形体之一,它由重复的碳五边形结构组成 [17]。一些研究预测 PG 在晶格常数固定的情况下是稳定的 [17, 18]。它具有蜂窝结构,是一种有前途的无金属、低成本的低温 CO 氧化催化剂 [19]。由于限速步骤的能量势垒非常小,氮掺杂的 PG 显示出非常高的催化活性,并且与许多金属基和碳基催化剂相比,在低温 CO 氧化方面更具竞争力[20]。也有报道称,过渡金属掺杂的 PG 是一种潜在的储氢材料 [21]。此外,与石墨烯不同,PG 是一种固有的准直接带隙半导体,带隙范围为 1.52-4.48 eV [8, 17, 22],这意味着在半导体气体传感器中的应用潜力巨大。此外,PG具有独特的杂化键结构,同时包含sp 3 和 sp 2 碳键。由于 sp 的四面体特性 3 由于碳键的杂化,PG不是理想的平面,而是以周期性波纹方式在平面外振荡[17],表明气体吸附作为传感元件的更多可能位置。
迄今为止,关于小气体分子与原始 PG 之间相互作用的研究很少。由于实验方法的限制,本研究采用密度泛函理论 (DFT) 计算来研究小气体分子(即 CO、H2O、H2S、NH3、SO2 和 NO)在新型活性炭上的吸附行为。碳材料PG。该研究将有助于工作人员分析和预测PG在气体传感器中的应用性能。
方法
在本研究中,结构优化的计算是通过基于 DFT [23] 的第一性原理计算进行的,如在 Dmol 3 中实施 代码 [24]。假设局部密度近似(LDA)有利于研究气体-分子-吸附系统[25, 26],本研究选择LDA-PWC进行结构优化。为了避免在研究气体-分子吸附时忽略范德华相互作用,采用了 Ortmann、Bechstedt 和 Schmidt [27] 的方法。 2 × 2 × 1 Monkhorst-Pack网格[28]用于布里渊区积分,其中自洽场容差设置为1 × 10 − 5 哈。当最大能量变化、最大力和最大位移的能量收敛精度为1 × 10 − 5 时,系统达到基态 Ha、0.002 Ha/Å 和 0.005 Å。进行了多核并行计算 [29] 并在 NO 吸附的计算中应用了自旋极化。基于包含两个 sp 的晶胞,对真空空间为 30 Å [21] 的 3 × 3 超胞进行建模 3 -杂化碳 (C1) 原子和四个 sp 2 -杂化碳 (C2) 原子 [17],见图 1,其中 C1 和 C2 原子分别被区分为黑色和灰色球体。气体分子以 3.5 Å 的初始距离与基板水平放置。为了获得气体分子最有利的吸附位置,研究了四个可能的位置,即 C1 原子的顶部 (T1)、C2 原子的顶部 (T2)、PG 凹槽的中间 (T3)、和T2的相反位置(T4),如图1所示。
<图片>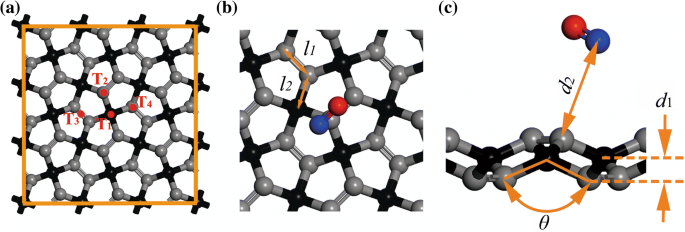
PG 的结构和几何形状:a 3 × 3个超胞,b 单元格的前视图,以及 c 吸附质-PG 原子的侧视图。两个C2原子之间的距离,C1和C2原子之间的距离,PG的厚度,C2-C1-C2的角度定义为l 1、l 2、d 1、θ , 分别
为了定量评价体系的吸附能力,除LDA-PWC外,吸附能(E a)、电荷转移(Q ), 吸附距离 (d 2,如图 1 所示)是在研究中使用 Perdew-Wang 1991 (PW91) 和 Perdew-Burke-Ernzerh (PBE) 使用广义梯度近似 (GGA) 函数计算的。 d 2 定义为平衡状态下 PG 与气体分子之间的最近原子距离。 问 表示气体分子 [30, 31] 的马利肯电荷,负值表示电荷从 PG 转移到气体分子。吸附能计算式为
$$ {E}_a\kern0.5em =\kern0.5em {E}_{\left(\mathrm{PG}\kern0.5em +\kern0.5em \mathrm{molecule}\right)}\kern0.5em -\kern0.5em {E}_{\mathrm{PG}}\kern0.5em -\kern0.5em {E}_{\mathrm{分子}},$$ (1)其中 E (PG + 分子), E PG 和 E 分子分别是吸附质-PG 平衡系统、分离的 PG 和分离的气体分子的总能量。在吸附系统电子结构的计算中,使用PBE交换相关函数和GGA的DFT计算已被用于更高的精度[2, 32]。
结果与讨论
结构优化后,本文报道的原始PG的计算结构参数(l 1 =1.342 Å, l 2 =1.551 Å, θ =133.9°,d 1 =0.612 Å) 被发现与之前的工作一致 [17]。通过选择最低的 E (PG + 分子)或E a 在 T1 到 T4 的四个吸附位置(参见附加文件 1 的表 S1),图 2 绘制了对气体在单层 PG 上最有利的吸附构型,以及最有利的吸附位置(即 T1、T2、T3 , 或 T4) 列于表 1 中。下文中的计算结果是基于这些最有利的吸附配置获得的。最近的研究表明,单层 InSe、石墨烯和蓝磷在气体传感器中具有广阔的应用前景 [14, 33, 34]。在本研究中,计算的 E CO、H2O 和 NH3 的 a 值分别为 - 0.531、- 0.900 和 - 1.069 eV(参见表 1),而对于 In,它们分别为 - 0.120、- 0.173 和 - 0.185 eV [33]。对于石墨烯表面的 CO、H2O、NH3 和 NO,计算的 E a 值分别为 - 0.014、- 0.047、- 0.031 和 - 0.029 eV [14]。同时,E PG 上 H2S 和 SO2 气体的 a 值分别为 - 1.345 和 - 1.212 eV,分别远大于蓝磷的 - 0.14 和 - 0.20 eV [34]。显然,计算出的 E 这些气体分子在 PG 上的 a 值远大于从其他材料获得的值,表明这些气体分子很容易吸附在 PG 表面 [35]。考虑到计算出的 E 一氧化碳的值远小于其他气体,对一氧化碳的吸附可能是最弱的。同时,表 1 中 NO 的吸附能小于一些物理吸附(非共价)吸附物,如 H2S、NH3 和 SO2。这可以解释为与物理吸附不同,NO的化学吸附引起PG的明显变形,消耗了额外的能量并降低了计算吸附能E a,如附加文件1中所述。同样,NO2在锑烯表面的化学吸附也可以观察到明显的变形[5],这可能导致吸附能相对较低。此外,除了 NO,d 的值 表1所列的2明显大于气体分子中相应原子与PG中C原子的共价半径之和(即l C-O =1.38 Å, l C-H =1.07 Å, l C-N =1.46 Å, l C-S =1.78 Å) [36],表明这些气体分子倾向于被物理吸附。关于 NO,d 的值 吸附体系中的 2 为 1.541 Å,处于共价键范围内,说明此时可能存在化学键。
<图片>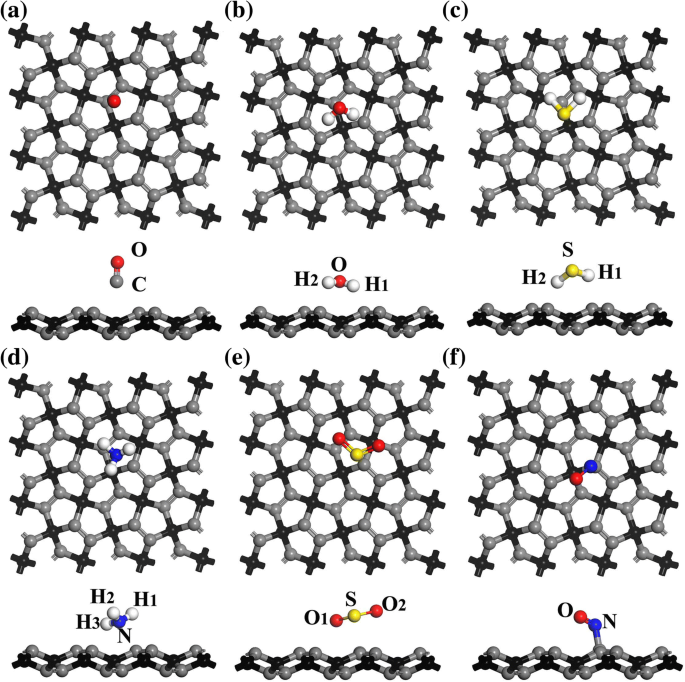
a 最有利吸附配置的俯视图和侧视图 CO,b 水,c H2S,d NH3,e SO2 和 f PG 上没有。灰色、红色、白色、黄色和蓝色球体分别代表C、O、H、S和N原子
先前对 InSe 和磷化硼的研究表明,吸附分子通过充当电荷受体或供体 [33, 37] 来改变基材的电阻率。 Q PG 表面 CO、H2O、H2S、NH3、SO2 和 NO 的值分别为 0.023、0.082、0.133、0.169、- 0.109 和 - 0.03 e(见表 1),表明 CO、H2O 、H2S 和 NH3 向 PG 提供电子,而 SO2 和 NO 从 PG 获得电子。值得一提的是,Q InSe 表面 CO、H2O、NH3 和 NO 的值分别为 0.006、0.014、− 0.025 和 0.018 e [33],石墨烯表面 CO、H2O、NH3 和 NO 的值分别为 0.012 、- 0.025、0.027 和 0.018 e 分别 [14],表明 PG 上气体分子的电子得失比 InSe 和石墨烯更明显。此外,虽然NO和PG之间可能发生化学吸附,但电荷转移仅为- 0.030 e。这可以通过以下事实来解释:N 和 O 原子的马利肯电荷分布在化学吸附之前和之后完全不同(参见附加文件 1 中的表 S2)。 O原子的增益电子抵消了N原子的损失电子,导致NO的总电荷转移量不大,而NO与PG之间的电子相互作用仍然明显。根据InSe和InN表面小气体分子电荷转移的传感机制[33, 38],推测PG在基于电荷转移机制的气体传感器中具有很大的应用潜力。
此外,还使用两个 GGA 泛函 PW91 和 PBE 计算了 PG 上的气体吸附。 E 的计算值 一、Q , 和 d 表 2 中列出了 PG 上的气体分子。E PW91 和 PBE 计算的 a 值小于 LDA 计算的值,而 PW91 和 PBE 均给出更大的 d 2 与 LDA 相比。与 LDA 不同,GGA 通常有低估吸附能和高估键距的倾向,这与先前工作的结果一致 [26, 31]。值得一提的是,这三个泛函的结果趋势是一致的。例如,E 的计算值 CO 的 a 最小,E 的计算值 H2S 和 SO2 的 a 大于其他气体分子的 a。此外,d 的计算值 LDA、PW91 和 PBE 泛函的 NO 2 分别为 1.514、1.592 和 1.591 Å,均在共价键范围内 [36]。
为了更好地理解气体分子对PG电子性质的影响,计算了分子-PG系统的态密度(DOS),见图3。显然,在费米能级附近(E f,例如,在- 2.5 到 2.5 eV 的范围内),H2O、H2S、NH3、SO2 和 NO 的电子能级对吸附系统有明显的贡献,表明这些气体分子的存在可能具有PG 的电子特性有很大影响 [5, 37]。例如,对于 H2S,电子能级的明显贡献位于 0 eV;参见图 3c。对于PG表面的CO,吸附系统中CO的轨道峰位于- 8.0、- 5.7、- 2.9和4.0 eV,在E附近没有明显的轨道贡献 F。此外,带隙也是决定材料电子特性的关键因素[26, 34]。
<图片>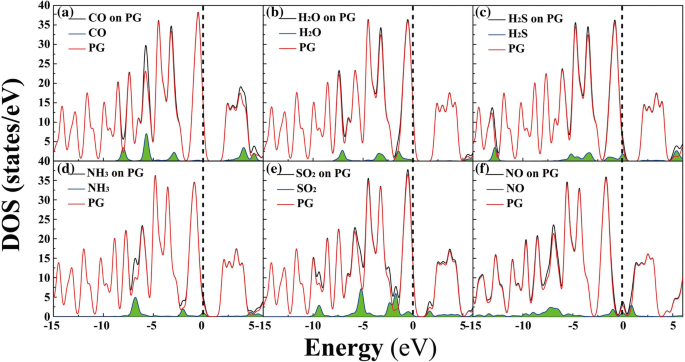
分子-PG 系统(黑色)的总电子态密度(DOS),以及吸附系统中小分子(带绿色阴影的蓝线)和 PG(红色)的投影 DOS:a CO,b 水,c H2S,d NH3,e SO2 和 f 不。费米能级设置为零(见虚线)
吸附系统的带隙和相应的带结构如图 4 所示,其中 CO、H2O、H2S、NH3、SO2 和 NO 在 PG 上的带隙分别为 2.15、2.02、1.86、1.81、1.61 和 0 eV,分别。相比之下,原始 PG 的带隙为 2.21 eV(参见附加文件 1:图 S2)。显然,除 CO 外,H2O、H2S、NH3、SO2 和 NO 的气体吸附对 PG 的电子性质有明显影响,这与 DOS 的结果一致。所有这些结果可能表明,除CO外,吸附H2O、H2S、NH3、SO2和NO后,PG的电子性质可以有效改变,这对气体检测至关重要。
<图片>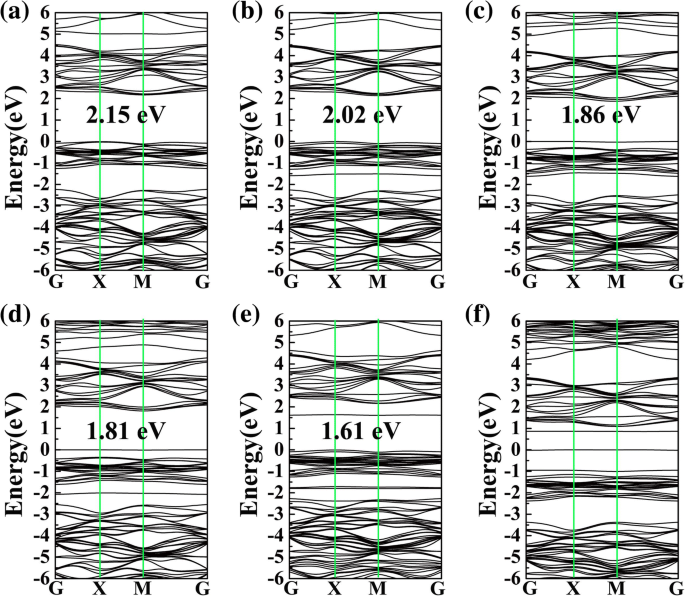
CO的能带结构 (a ), H2O (b ), H2S (c ), NH3 (d ), SO2 (e ) 和否 (f )在PG表面
考虑到 d 2 PG上NO的值在键合范围内,系统中NO的电子能级主要集中在E附近 f,推测发生了化学吸附。为了深入了解 NO 和 PG 之间的吸附机制,图 5 绘制了 NO 在 PG 上的投影态密度 (PDOS) 和电子局域函数 (ELF)。显然,N 的 PDOS 的峰值 p 和 O p 原子主要位于 - 6.9、- 0.9、0 和 0.8 eV;因此在 NO 中存在分子内杂交,如图 5a 所示。同时,在费米能级附近可以观察到 NO 和 PG 的 C 原子之间的轨道混合,这主要是由 C s 贡献的 , C p , N s , N p , 和 O p 轨道。轨道混合导致 NO 中的 N 和 PG 中的 C 之间形成化学键,如图 5b 所示;因此,PG 可用于检测或催化 NO 气体 [5, 39]。此外,为了确认其他气体分子的吸附类型,还计算了其他吸附系统的 ELF,参见附加文件 1:图 S3。显然,对于CO、H2O、H2S、NH3和SO2,气体分子与底物之间不存在化学键,表明体系趋向于物理吸附。
<图片>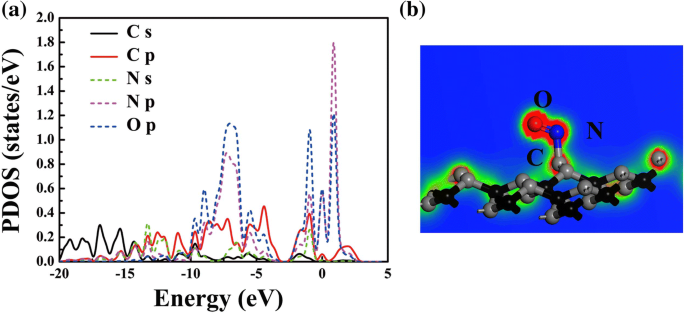
原子投射 DOS (a ) 和 NO-PG (b )
结论
总之,H2O、H2S、NH3 和 SO2 气体物理吸附在单层 PG 上,具有相当大的吸附能和适度的电荷转移。由于物理吸附弱,吸附能小,电荷转移,原始PG不适合检测CO。对于PG表面的这些气体分子,CO、H2O和H2S,NH3给PG提供电子,而SO2和NO获得PG 的电子。此外,在费米能级附近,H2O、H2S、NH3、SO2 和 NO 的电子能级对吸附系统的 DOS 有明显贡献,表明在 H2O、H2S、 NH3、SO2 和 NO 被吸附。此外,PG对NO的吸附表现出强烈的化学吸附趋势,因此PG可用于检测或催化NO气体。因此,Pristine PG在气敏应用中具有巨大的潜力。
缩写
- 二维:
-
二维
- DFT:
-
密度泛函理论
- DOS:
-
态密度
- ELF:
-
电子定位功能
- GGA:
-
广义梯度逼近
- LDA:
-
局部密度近似
- PBE:
-
Perdew-Burke-Ernzerh
- PDOS:
-
状态的投影密度
- PG:
-
五石墨烯
- PW91:
-
Perdew-Wang 1991
- PWC:
-
Perdew-Wang 相关
纳米材料