

CH4 气体分子在 MoX2(S、Se、Te)单层上的吸附行为:DFT 研究
摘要
我们通过密度泛函理论 (DFT) 预测具有 X 空位、Mo 空位和双空位的单层 MoX2(S、Se、Te) 的 CH4 传感性能。结果表明,不同的第六主族元素与Mo原子的结合对CH4气体分子具有不同的吸附行为。与 MoX2 相比,MVX、MVMo 和 MVD 在相同条件下通常表现出更好的吸附性能。此外,不同的缺陷会对系统的吸附行为产生不同的影响,MVD(MoTe2)在这些系统中具有更好的吸附性、更好的电荷转移和最短的距离。该结果可用于预测 MVD(MoTe2) 的 CH4 气体分子吸附特性,有助于指导实验人员开发更好的基于 MoX2 的材料,用于有效的气体检测或传感应用。
介绍
甲烷(CH4)是最简单的有机化合物,具有无色无味的气体[1,2,3,4],对人体基本无毒,当甲烷浓度过高时,空气中的氧含量会明显降低高,让人窒息。当空气中甲烷浓度达到25-30%时,会引起头痛、头晕、乏力、注意力不集中、呼吸心跳加快、共济失调[5,6,7]。自从石墨烯的兴起 [8, 9] 和拓扑绝缘体的发现 [10] 以来,在尺寸减小的系统中发现了许多有趣的物理现象。其他二维 (2D) 材料,例如过渡金属二硫属元素化物 (TMD) 的单层或少层系统(纳米层),因其固有带隙而变得越来越重要 [11,12,13,14,15]。 TMD 是 MX2 型化合物,其中 r (S, Se, Te) [16,17,18,19]。这些材料形成层状结构,其中不同的 X -M -X 层通过弱范德华力保持在一起 [20,21,22,23,24,25,26]。 Yi Li [27] 研究发现 COF2 在 Ni-MoS2 上的吸附能优于 CF4,并且 Ni-MoS2 作为电子供体,观察到明显的电荷转移。 Soumyajyoti Haldar [28] 报道了 2D 过渡金属二硫属化物 MX2 中原子尺度缺陷的结构、电子和磁特性,不同的空位对不同的 2D 二硫属化物 MX2 有很大影响,很可能是带隙、态密度、一些属性等等。 Janghwan Cha [29] 使用不同的泛函来显示气体分子和 MoX2 的相对结合能。 optPBE-vdW 泛函显示出相对较大的结合能。此外,TMDs 是实现气体传感器的有前途的材料,因此我们研究了许多缺陷对 MoX2(X=S, Se, Te) 的结构、带隙 [30,31,32]、吸附能、电荷转移、等。本文通过第一性原理模拟研究了甲烷与单层MoX2 的相互作用(见图1)。绿色球为 Mo 原子,黄色球为 X 原子,SS、Se-Se、Te-Te 的 d1 距离分别为 3.190 Å、3.332 Å 和 3.559 Å,d2 的距离为与d1的三种情况相同。本工作基于DFT,研究了CH4气体分子在MoX2上的吸附能、电荷转移、吸附距离和态密度(DOS)。
<图片>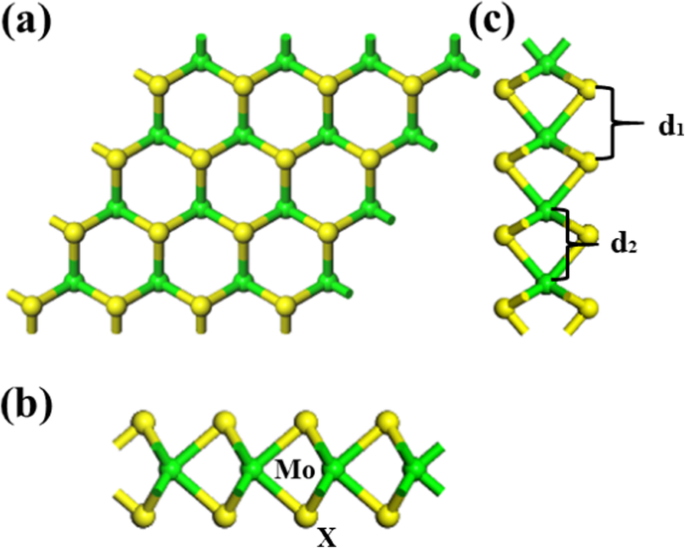
一 正视图。 b 侧面图。 c 左视图
方法与理论
在 Materials studio [33,34,35,36] 中构建了一个 4 × 4 的 MoX2(32 个 X 原子和 16 个 Mo 原子)和吸附在其上的 CH4 气体分子的超晶胞。 DMol 3 [37] 软件用于计算。在本文中,选择具有广义梯度近似 (GGA) 的 Perdew、Burke 和 Ernzerhof (PBE) [38, 39] 函数来描述交换能量 Vxc。 Mo 产生于 4p 6 5s 1 4d 5 配置和另一个用于生成 X 的价电子。使用 6 × 6 × 1 k 点网格和 0.01 Ry 的 Methfessel-Paxton 涂抹对 MoX2 的布里渊区进行采样。截止能量为 340 eV,自洽场 (SCF) 收敛为 1.0 × 10 −5 EV。所有原子结构都被松弛,直到最大位移容差为 0.001 Å,最大力容差为 0.03 eV/Å [40, 41]。
我们计算了吸附能 (E ad) 在吸附系统中,定义如下:
$$ {E}_{\mathrm{a}}={{E_{\mathrm{MoX}2+\mathrm{CH}4\ \mathrm{gas}}}_{\mathrm{m}}}_{ \mathrm{olecule}}-\left({E}_{\mathrm{MoX}2}+{E}_{\mathrm{CH}4\ \mathrm{gas}\ \mathrm{molecule}}\right) $$其中,E MoX2 + CH4 气体分子,E MoX2 和 ECH4 气体分子分别代表单层 MoX2 吸附系统、单层 MoX2 和 CH4 气体分子的能量。所有能量在结构优化后达到最佳优化。我们使用Mulliken的人口分析来研究电荷转移。
结果与讨论
首先,我们讨论了四种 MoX2 衬底的几何和电学结构(见图 2)。 Mo-S、Mo-Se 和 Mo-Te 的键长分别为 2.426 Å、2.560 Å 和 2.759 Å,与实验值 2.410 Å (MoS2) [42, 43]、2.570 Å ( MoSe2) [44] 和 2.764 Å (MoTe2) [45],本文中的四种结构 MoX2,原始 MoX2、MVX(一个 X 原子空位)、MVMo(一个 Mo 原子空位)和 MVD(一个 X 原子和一个 Mo 原子空位)分别。完全结构松弛表明 X-Mo 键长从 2.420 Å 拉伸到 2.394 Å (MVS),2.420 Å 到 2.398 Å (MVMo),主要原因是原子的缺失增强了相邻 Mo 原子和其他S原子,化学键变强,键长变短。
<图片>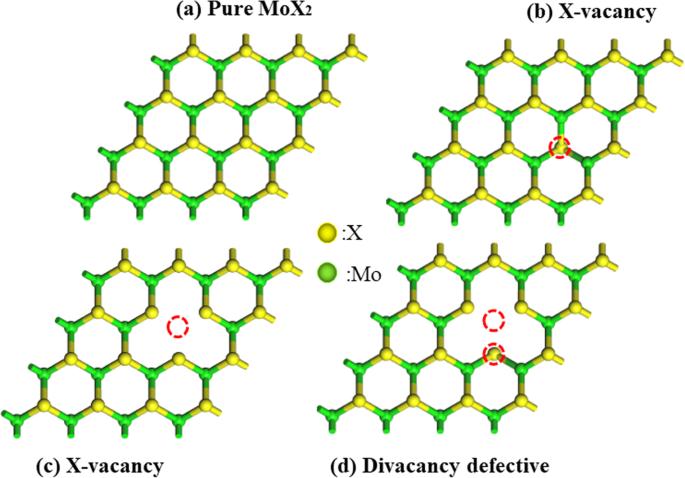
带有 a 的 MoX2 顶视图 纯 MoX2,b S 空缺,c Mo 空缺和 d 空缺。绿色和黄色球分别代表Mo和X(S, Se, Te)原子。
图 3a-c 显示了计算的 CH4/MoX2 系统的吸附能、电荷转移和吸附距离。在吸附之前,CH4 气体分子与二硫化钼之间的距离为 3.6 Å。 CH4气体分子从MoS2片的四个系统获得约- 0.001e到- 0.009e,从MoS2片的四个系统得到- 0.009e到- 0.013e,从MoS2片的四个系统得到- 0.014e到- 0.032e ,分别表示 CH4 作为受体。包含范德华修正后,CH4 气体分子的吸附能在四个 MoS2 系统上提高了 - 0.31 eV 到 - 0.46 eV,在四个 MoSe2 系统上增加了 - 0.07 eV 到 - 0.50 eV,以及 - MoTe2 系统的四个系统上的 0.30 eV 到 - 0.52 eV,通常认为 0.01 eV 在误差范围内。很明显,在 S 原子缺陷和双空位缺陷的情况下,吸附距离最短。综合以上数据可知,双空位缺陷条件下吸附效果最好。
<图片>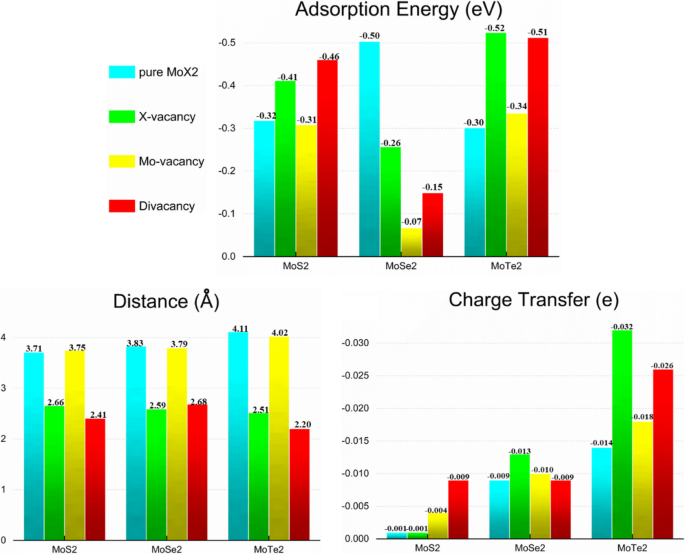
吸附能、分子与MoX2之间的最短原子距离和电荷转移
CH4气体分子在单层MoS2上的吸附
为了清楚地了解 CH4 气体分子在纯和有缺陷的 MoS2(包括 MV、MVMo 和 MVD)上的键合机制,我们分析了吸附结构中吸附的 CH4 气体分子的相应态密度(DOS)。比较四个系统,进一步研究了 CH4 气体分子对纯和有缺陷的 MoS2(包括 MV、MVMo 和 MVD)的吸附效果。 DOS(图4)显示费米能级附近有一定的变化,与一般的DOS形式相同。沿伽马点 (G) 观察到四个系统的能带隙,注意到为 1.940 eV (MoS2)、1.038 eV (MVS)、0.234 eV (MVMo) 和 0.209 eV (MVD)。此外,观察到的 MoS2 纳米片的能带隙与其他报道的理论工作(1.78 eV [39]、1.80 eV [40])和实验工作(1.90 eV [41]、1.98 eV [42])非常吻合。同时,单层MoS2有五个峰值,峰值分别为- 12.2 eV、- 5 eV、- 4 eV、- 2 eV和- 1 eV,它们分别归因于MoS2中的S原子和MoS2中的Mo原子。然而,四个系统的 DOS(图 4)显示 CH4 气体分子的电子能级在约 - 3 eV 处有一个峰值,该峰值接近于费米能级。它有助于系统中的导带并影响系统的电导率。比较四个系统,由于 MoS2 中 S 原子的缺陷,− 12.5 eV MVs 的峰值明显低于 MoS2。而Mo原子的缺陷影响不大;然而,传导区的贡献仍在下降。如图3b所示,很明显,0 eV附近的能带越来越小,曲线越来越稳定。综上所述,CH4气体分子与MoS2无键,电子转移和吸附能小,吸附力不强,明显是物理吸附。
<图片>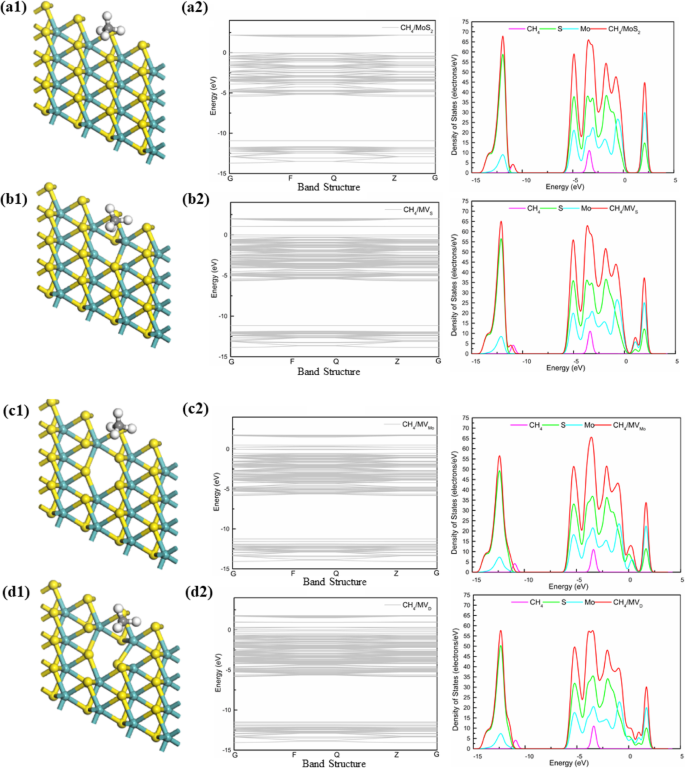
四种体系(MoS2、MVS、MVMo和MVD)上CH4气体分子的结构和DOS
CH4 气体分子在单层 MoSe2 上的吸附
我们研究了 CH4 气体分子在 MoSe2 的四个系统上的吸附,从 DOS(图 5)可以看出,CH4 气体分子在四个吸附方向的电子能级接近费米能级,具有对体系的电导率有一定影响,带隙体系很小,与吸附 MoS2 相同。同时,DOS(图5)还显示MoSe2中的Se原子有五个峰值,峰值为- 12 eV、- 5 eV、- 4 eV、- 3 eV和- 2 eV,MoSe2中的Mo原子MoSe2 在约 0.5 eV 和 2 eV 处有重叠峰。与 MoS2 相比,在费米能级以下的 MoS2 中,Se 对系统的贡献大于 S,并且沿伽马点 (G) 观察到四个系统的能带隙,注意到 1.680 eV (MoSe2)、1.005 eV ( MVSe)、0.094 eV (MVMo) 和 0.024 eV(MVD)。该带在 0 eV 附近更窄且更稳定。因此,可以确定4个体系的吸附性质和CH4气体分子均为物理吸附。
<图片>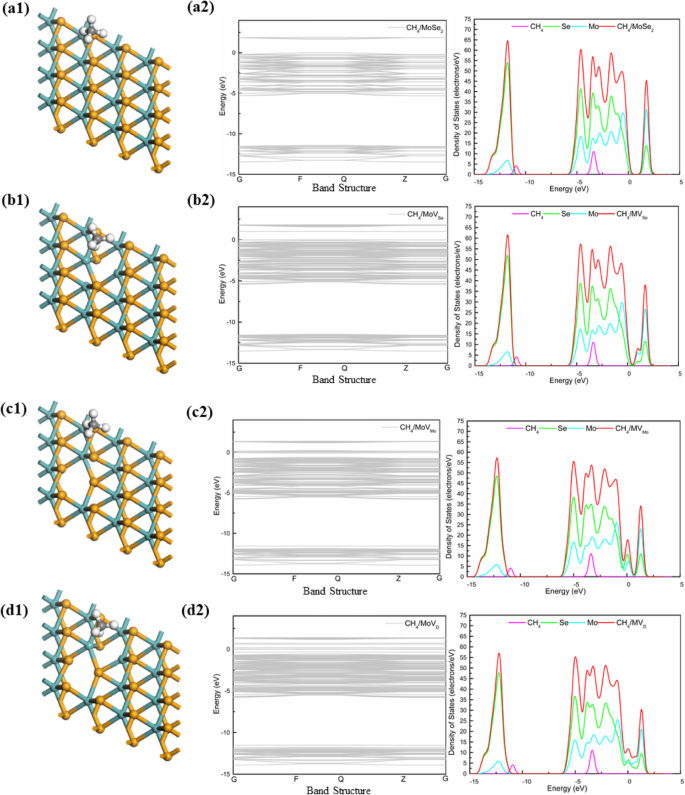
四种体系(MoSe2、MVSe、MVMo和MVD)上CH4气体分子的结构和DOS
CH4气体分子在单层MoTe2上的吸附
我们研究了 CH4 气体分子在 MoTe2 的四个系统上的吸附,分析了 CH4 气体分子在 MoTe2 上的 DOS(图 6)。如图 6 所示,四种 MoTe2 体系中 CH4 的电子能级与 CH4/MoS2 体系和 CH4/MoSe2 体系短,沿伽马点(G)观察到四种体系的能带隙为 1.261 eV (MoTe2)、0.852 eV (MVTe)、0 eV (MVMo) 和 0.316 eV (MVD)。最奇怪的事情之一是钼原子的缺陷,它允许系统从半导体转变为金属。同时,DOS(图6)还显示MoTe2中的Te原子有四个峰值,峰值为- 10 eV、- 5 eV、- 3 eV和- 1 eV,MoSe2中的Mo原子有重叠峰约 1 eV。
<图片>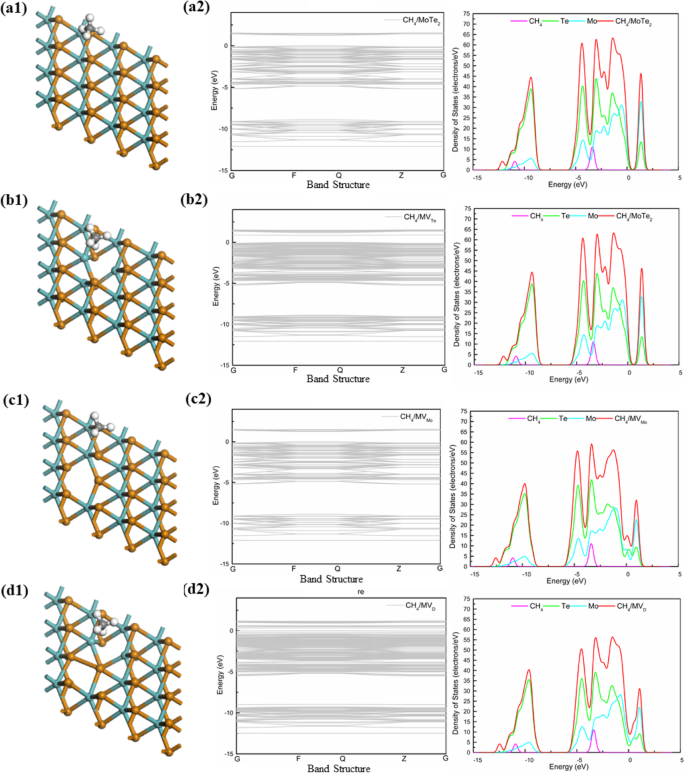
四种体系(MoTe2、MVTe、MVMo和MVD)上CH4气体分子的结构和DOS
一般来说,根据CH4气体分子在不同体系中的吸附行为,MVX吸附的CH4气体分子在费米能级附近可能有两个峰。两个尖峰之间的 DOS 不是零而是很宽,这反映了系统的强共价特性。综上所述,MVTe可能成为检测CH4气体分子的理想传感材料。
结论
我们进行了密度泛函 GGA 研究,以研究孤立的 CH4 气体分子在 MoX2(X=S、Se、Te)上的相互作用。结果表明,不同的缺陷极大地改变了 MoX2 的电学性质,我们的结果揭示了 CH4 气体分子与 MoX2 单层之间的弱相互作用,这表明吸附的物理性质。总电子密度图证实了 MoX2 表面上的气体分子的物理吸附,因为该材料与 CH4 气体分子的相互作用很弱,而在界面区域没有形成共价键。此外,MVD的结构具有良好的带隙、半导体性质、最佳的吸附能以及对CH4气体分子更强的电荷转移。此外,传感系统的电子能带结构随着气体分子的吸附而改变。 MoTe2 具有最高的吸附能(- 0.51 eV)、最短的分子间距离(2.20 Å)和更高的电荷转移(- 0.026 e)。最后从这三种材料的分析可以看出,MVD(MoTe2)对CH4气体分子的吸附效果最好。计算结果为MVD(MoTe2)单分子层在CH4基气体传感器器件中的潜在应用提供了理论基础。
数据和材料的可用性
所有数据完全可用,不受限制。
缩写
- CH4:
-
甲烷
- DOS:
-
态密度
- Ea:
-
吸附能
纳米材料