

直接还原过程的理论方面
直接还原过程的理论方面
在铁矿石的直接还原过程中,固体金属铁(Fe)是直接从固体铁矿石中获得的,无需使矿石或金属熔融。直接还原可以定义为在氧 (O2) 电位下的固态还原,这允许氧化铁还原成相应的元素,但不能还原其他氧化物(MnO、SiO2 等)。由于还原处于固态,这些元素很少有机会溶解(在低热力学活性下)在还原铁中,因此比铁更稳定的氧化物基本上保持未还原。高炉竖井中的铁矿石也被上升的气体直接还原。
铁氧系统
铁氧(Fe-O)系统可能是研究最广泛的系统之一。该系统的热力学是众所周知的,并且有很多关于涉及氧化铁的气体还原动力学的信息。在总压力为 I kg/sq cm 的 Fe-O 系统中,在 400 摄氏度和 1400 摄氏度之间出现的热力学稳定的固相如图 1 所示。该图显示 Fe 与 O2 形成三种稳定的固体化合物,即 (i) 赤铁矿 - Fe2O3,(ii) 磁铁矿 - Fe3O4 和方铁矿 - FexO,其中 x 略低于 1。非化学计量的 FeO 相 (方晶石)在 570 摄氏度以下不稳定,分解成金属 Fe 和 Fe3O4 的混合物。因此,在恒温下从右到左读取相图,低于 570 摄氏度的相序为 Fe2O3 - Fe3O4 - Fe,而高于 570 摄氏度的相序为 Fe2O3 - Fe3O4 - FeO - Fe。
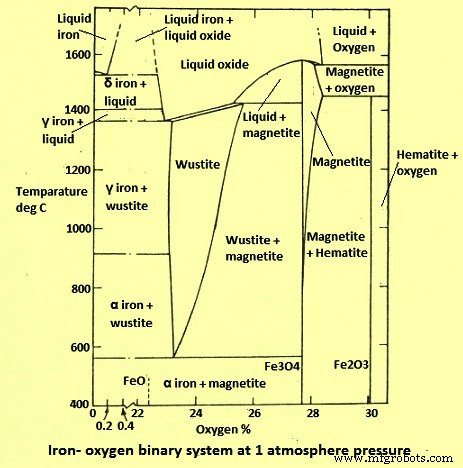
图1 Fe-O二元系统图
O2 在固体 α 和 γ 铁中的溶解度可忽略不计,小于 O2 的 0.01%。因此,O2 含量对固体 Fe 改性的转变温度没有影响,在图中被忽略了。
考虑到反应平衡,氧化铁的还原涉及以下步骤中的一个或多个:(i) 赤铁矿 (Fe2O3) -> 磁铁矿 (Fe3O4),(ii) 磁铁矿 (Fe3O4) -> 铁 (Fe),(iii) 磁铁矿 ( Fe3O4) -> 方晶石 (FeO),和 (iv) 方晶石 (FeO) -> 铁 (Fe)。
方晶石仅在高于 570 摄氏度的温度下才稳定。上述反应的热力学平衡因使用的两种主要气态还原剂氢气 (H2) 和一氧化碳 (CO) 而众所周知。
铁-氧-碳系统
Fe和Fe氧化物与CO和CO2(二氧化碳)气体与固体碳(C)的混合物的平衡如图2所示。
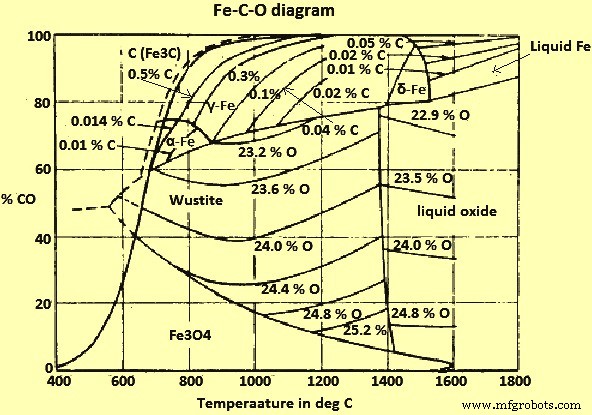
图2 Fe-CO-O系统图
从图 2 可以推断,在 710 摄氏度以上的温度和 1 kg/sq cm 的总压力下,所有的 Fe 氧化物都可以被与 C 平衡的 CO/CO2 气体混合物还原,并且可以,因此,由 C 本身减少。在较低温度下,只有那些 C 过饱和的混合物,因此,根据 Boudouard 平衡,对 C 沉积起反应的混合物对方晶石具有还原作用。
铁-氢-氧系统
Fe和Fe氧化物与H2和H2O(蒸汽)混合气体的平衡图如图3所示。
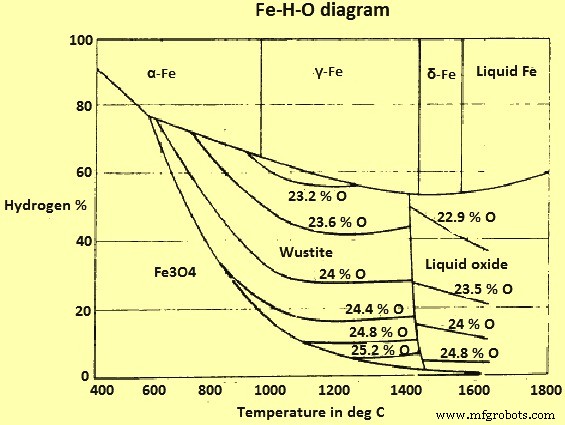
图3 Fe-H-O系统图
该系统与 Fe-O-C 系统的主要区别在于没有“烟灰线”或相应的现象。因此理论上可以在任何温度下用H2将赤铁矿(和磁铁矿)还原为Fe。
CO和H2还原的比较
根据对 Fe-C-O 和 Fe-H-O 系统的研究(图 4),似乎在 815 摄氏度以上,H2 是比 CO 更有效的还原剂(即平衡 H2/H2O 比低于相应的 CO /CO2 比率),而在较低温度下则相反。然而,这些平衡在工业炉中很难实现,因为随着接近平衡,还原速率变得非常慢。当条件明显偏离平衡时,用 H2 和 CO 还原的相应反应速率与通常从平衡考虑中预期的反应速率相反。因此,对于非平衡过程而言,H2 实际上是一种更有效的还原剂,该过程被设计为在低于 815 摄氏度的温度下运行,而 CO 在更高的温度下更有效。
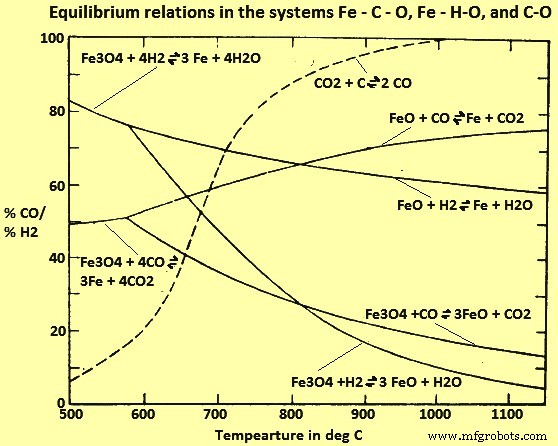
图 4 Fe-C-O、Fe-H-O 和 C-O 体系中的平衡关系
对不同温度下 CO 和 H2 气体组成混合物影响的研究表明,随着还原气体混合物中 H2 含量的增加,反应速率会增加。已经发现这种关系是显着非线性的。
用 H2 将 Fe 氧化物还原为金属 Fe 是吸热的,需要外部热源来维持所需的温度。与 CO 的相应反应是放热的,并且在适当控制的条件下,该反应是热自持的。事实上,可能需要用 H2 或其他吸热气体稀释 CO,以避免装料过热。一些工艺旨在利用 CO - H2 热平衡,并使用这些气体的混合物来增加矿石加热过程中获得的还原量,从环境温度到最高反应温度约 1100 摄氏度。
图 4 表明,对于在经济可行的气体还原范围内的所有温度,平衡气体混合物包含至少 60% 的 CO 和/或 H2。当未达到平衡时,这些气体的未反应浓度甚至更高,并且大部分不变地通过还原炉。如果该方法是经济的,那么有必要利用在方晶石还原成金属Fe之后剩余的气体,用于将较高的Fe氧化物还原成方晶石和/或用于气体混合物的再生和气态反应产物的去除。
气固反应和固固反应
气固反应在技术中发挥着重要作用,涵盖非常广泛的领域,包括从矿石中提取金属(氧化铁还原等)。所有气固反应系统的一个共同特点是整个过程可能涉及多个中间步骤。通常,这些中间步骤包括 (i) 反应物和产物从大部分气相到反应固体颗粒的外表面的气体扩散(传质), (ii) 气态反应物或气态产物通过气孔的孔扩散。固体反应产物或通过部分反应固体的孔隙,(iii) 气态反应物在固体表面的吸附和反应产物从固体表面的解吸,以及 (iv) 吸附气体和固体之间的实际化学反应。 /P>
在气固反应领域,还有一些其他现象会影响反应的进程和进行气固反应的炉子的性能。这些其他现象包括热传递、伴随反应的固体结构的变化(如烧结),以及气体和固体通过发生气固反应的熔炉的流动。还原率由这些因素控制,具体取决于所使用的工艺。
固体之间的反应可分为两大类,即(i)真正的固体——固体反应发生在两个相互接触的颗粒之间,或通过固体颗粒的迁移(例如形成通过 Fe 氧化物和 C 之间的反应来还原碳化铁,以及 (ii) 固体反应物之间的反应,这些反应通过气态中间体发生(例如在 1 kg/sq cm 压力下用碳还原 Fe 氧化物)。
用固体 C 还原铁氧化物也可以是真正的固-固反应,只要它在非常低的绝对压力下进行。在一项由细粉石墨 (C) 和赤铁矿的混合物在 0.0005 毫米汞柱(汞)的真空下进行的研究中发现,在高达 900 摄氏度的温度下,反应发生得非常快。慢慢地,在 18 小时内只有 Fe3O4 和 FeO 形成,但没有 Fe。在测试期间,仅在较高温度下观察到明显的转变。研究期间得出的结论是,反应速率取决于 Fe 离子在氧化物相中的扩散。在另一项研究中作出的推论是 C 扩散在氧化铁中,这可能只是具有历史意义。然而,这些研究表明,当粉末混合物上方的气压增加时,反应速率明显增加。在类似类型的试验中,其中 N2(氮气)流通过 C 和 Fe 氧化物的混合物,随着 N2 流量的增加,观察到反应速率显着降低。所有这些研究,无论是在真空中还是在 N2 下,以类似粉末状 Fe 氧化物在 CO 或 H2 中的还原速率,证明了 C 和矿石的直接固态反应(有时被认为是实际机理)真正的直接还原)对工业炉还原过程的进展并不重要。
还原铁的孔结构
对几种天然矿石的还原性试验表明,铁矿石颗粒的孔隙率是控制还原性的最重要因素之一。还原性表示为 90% 还原所需时间的倒数,直接随孔隙率而变化。相对还原率随着孔隙率的增加而增加,由公式“相对还原率 =(孔隙率 x 0.75) + 8.0”给出。
Fe氧化物的还原总是产生多孔反应产物。氧化物的性质和还原条件影响还原铁的孔结构。这是因为还原是从颗粒表面向内进行的。由原始方晶石表面定义的体积占据空间减少了。这只能通过增加孔隙率来实现。对该孔隙度的扫描电子显微照片研究表明,一般而言,H2 还原得到的孔结构比 CO 还原得到的孔结构更细。另外,从扫描电子显微照片可以看出,随着氢气还原温度从600℃逐渐升高到1200℃,孔结构变得越来越粗。
Fe氧化物的初始孔表面积影响通过气体还原形成的还原铁的孔表面积。 Fe氧化物的初始孔表面积的减少减少了还原铁的孔表面积。用BET(Brunauer-Emmett-Teller)技术测定赤铁矿在氢气中还原的铁的孔表面积随着还原温度的升高而减小。
从孔径分布中得到了还原温度与平均临界孔径和最小孔径之间的关系。已发现孔径随着还原温度高达 900 摄氏度而缓慢增加,但随着温度的进一步升高而迅速增加。这些结果与扫描电镜观察断口一致,在900℃以上的还原温度下,孔隙结构明显粗化。
还原后的赤铁矿的孔表面积还受还原温度和气体成分的影响。赤铁矿在 CO/CO2 气体混合物中还原得到的孔表面积约为 H2/H2O 气体混合物还原所得孔表面积的三分之二。这与显微镜下观察到的CO/CO2还原铁的较粗孔结构一致。
已测量还原铁孔隙中的气体扩散。多孔介质中的扩散通量通过两个扩散过程发生,即(i)克努森扩散,与压力无关,与 T(温度)成 1/2 次方成正比,以及(ii)分子扩散,与压力成反比,与 T 成正比功率3/2。
假设极限理想结构具有大小均匀的孔洞,这些孔洞都相互连通,并以45度角相交。
对于给定的多孔介质,有效扩散率随温度和压力而变化,并且对于不同的二元气体对也不同。随着还原温度的降低,孔隙结构变细。
减少方式
天然铁矿石颗粒或烧结赤铁矿球团的还原导致产品层的形成。这种众所周知的现象是许多研究的主题。在最近的一项关于 H2 还原烧结赤铁矿球团的研究中,已经注意到在部分还原的赤铁矿球团的抛光部分存在层形成的典型例子。层之间相对光滑的界面通常在低放大倍率下出现,尽管这样的外观可能会产生误导。
这表明在方晶石层中的气体扩散足以在前进的 Fe/FeO 界面之前产生一些内部还原。随着(i)温度降低,(ii)孔隙率提高,(iii)粒径变小,内部还原区扩大。
粒度对达到给定百分比还原所需时间的影响取决于还原模式,因此取决于速率控制过程的类型。考虑通过气体还原来还原多孔铁氧化物的模式已经显示出三种限制速率控制过程,即(i)均匀内部还原,(ii)限制混合控制,和(iii)多孔铁层中的扩散。如果还原仅由其中任何一种控制,则还原时间以三种方式之一与颗粒(球形)直径相关,即(i)均匀内部还原,即时间与直径无关,(ii)限制混合控制,以及 (iii) 在多孔铁中的扩散。
仅当 (i) 存在均匀的内部还原,因此需要小粒径,或 (ii) 通过铁层孔隙中的气体扩散来控制最终速率占主导地位时,速率控制过程才变得相对简单,因为粒径很大。还应认识到,随着还原的进行,可能存在从一种限制速率控制过程到另一种的转变,这取决于温度、气体成分、颗粒大小和氧化物的类型。氧化铁的还原也会表现出一些无法解释和不寻常的行为。
多孔铁矿颗粒的还原率
矿石的孔隙度和孔隙结构对内部还原的程度和均匀性有显着影响。在其中一项研究中,对于 90% CO 和 10% CO2 的混合物以及 1000 摄氏度的 H2,粒度对赤铁矿还原率的影响表明,随着粒度的增加,内部还原受到限制到颗粒的外部区域,因此随着颗粒尺寸的增加,整体还原率降低。
在多孔赤铁矿颗粒还原的早期阶段,快速转化为 FeO,随后 FeO 内部还原为 Fe。在氧化铁的孔中几乎完美的气体扩散的极限情况下,内部还原占主导地位,并且速率主要由孔壁上的气固反应控制。假设几个原子厚的 Fe 层覆盖了 FeO 的孔壁。还原速率被认为是由 O2 通过孔壁上的 Fe 层涂层的快速扩散以及 H2 或 CO 与这个非常薄的 Fe 层表面上的 O2 的化学反应共同控制的。
颗粒大小的影响表明,还原速率随着颗粒大小的减小而增加。典型的显微照片表明,还原模式在颗粒内从一个晶粒到另一个晶粒变化。这是因为氧化物晶粒孔隙率的局部差异。由于孔径的变化和较大孔隙中气体扩散较快,大部分反应发生在较大孔隙的壁上。也就是说,预计只有一小部分总孔表面积用于反应。用各种类型的赤铁矿颗粒在 800 摄氏度下实现的 H2 还原速率随着所形成的 Fe(或 FeO)的孔表面积的增加而非线性增加。这些结果证实了孔表面积越大,反应中使用的总孔壁的比例越小。
H2-CO 气体混合物的内部还原速率通常是 H2 和 CO 的两个单独还原速率的总和。还原数据和观察到的 C 沉积表明,在 1000 摄氏度以下,气体反应导致水-气体平衡很慢。
铁矿石(块状或球状)的还原率
块矿或球团矿的还原速率在填充床中的还原气流中具有复杂的性质,其复杂性是因为总体还原速率由几个串联的反应过程控制,例如热量和质量通过气膜边界层传递、气固反应和多孔产品层中的气体扩散。通过数学分析,借助计算机计算,推导出了许多方程来描述不同还原模式下大氧化物颗粒的还原速率。
在几个使用单个球团或铁矿石颗粒的实验中,传热相对较快,并且在气流速度足够高的情况下,气膜传质阻力小到可以忽略不计。因此,主要有两个主要的串联反应步骤会影响还原速率,即 (i) 气体-氧化物反应,和 (ii) 多孔氧化物和多孔产物层中的气体扩散。这些速率过程的相对影响取决于颗粒大小、气体成分、温度和还原方式,并随着还原的进行而变化。
多孔铁层中的气体扩散
在其中一项研究中,进行了单向还原实验以证明气体扩散在 Fe 层孔隙中的影响。长圆柱样品由大块赤铁矿块制成,并装入紧密配合的镍管内。在 H2 还原所需的时间后,将样品轴向分割并抛光,并确定 Fe 层的厚度。实验结果表明,当还原铁层的厚度在 1 mm 左右时,进一步还原符合抛物线速率规律,这与孔隙扩散控制的结果相似。这些试验表明,随着多孔Fe层厚度的增加,还原速率最终受Fe层孔隙中的气体扩散控制。
在 Fe 层的主要前进前沿之前的部分内部还原会导致还原层中截留一些 FeO。这种情况会导致还原最后阶段的 O2 去除缓慢。
随着还原温度的降低,孔结构变得更精细,可能在连接的毛细管上有许多狭窄的通道和瓶颈,当克努森扩散占主导地位时,有效分子扩散率/有效平均克努森扩散率的比率值较低。随着还原温度的升高,孔隙结构变粗,气体容易通过孔隙,该比值变高。
气体组成对烧结赤铁矿球团和磁铁矿球团在 900 摄氏度下通过 H2-CO-CO2 混合物(具有 CO/CO2 比等于 9 以抑制烟灰沉积),是当 H2 被 CO 取代时,等温还原达到给定百分比的 O2 去除的时间逐渐增加,直至约 50% CO,并且随着进一步添加 CO,在 CO 中显着增加减少的时间。 100%(CO/CO2 比率等于 9)的还原时间大约是相同温度下 H2 的 10 倍。从气体动力学理论推导出的二元系统(例如 H2-H2O 或 CO-CO2)中的分子气体扩散率对于系统而言是不变量,并且基本上与气体成分无关。然而,在三元和多组分系统中,每种物质具有不同的扩散率,并随气体成分而变化。此外,扩散通量的速率方程很复杂。
赤铁矿球团在 H2-CO 混合物中的还原行为显示出类似于在 H2 和 CO 中观察到的模式,即超过约 50% O2 去除的还原速率受 Fe 孔隙中的气体扩散控制层。
在初始速率中限制混合控制
在还原的早期阶段,还原速率受以下因素共同控制:(i)FeO 孔隙中的气体扩散(FeO 中的固态扩散可以忽略),以及(ii)FeO 孔壁上的反应.这意味着薄的多孔铁层和其中的快速气体扩散。根据 FeO 的孔隙率和其中的气体扩散率,在标称 Fe/FeO 界面之前存在部分内部还原。 H2与多孔FeO的反应通常局限于靠近名义Fe/FeO界面的孔口。
部分内部减少
根据气体成分、温度、颗粒大小和总气体压力,在限制速率定律的框架内,在某些还原期间存在混合速率控制。速率方程通常基于这样的假设,即球团的气体还原是由气体通过球团颗粒间孔的缓慢逆流扩散以及气体与氧化铁的缓慢化学反应共同控制的。颗粒的Fe氧化物-/Fe界面。
水煤气变换反应
水煤气变换反应在使用重整烃作为还原剂还原氧化铁的直接还原过程中发挥着重要作用。一般认为,根据 CO 和 H2 对铁矿石的不同还原率,以及在 CO/CO2 混合物中含有少量 H2 对还原率的显着影响,H2 是实际的还原组分在这样的气体混合物中。 CO 被认为主要用于将生成的蒸汽 (H2O) 还原为 H2。反应为(i)H2 + FeO =H2O + Fe,和(ii)H2O + CO =H2 + CO2。
该反应的第二个子过程称为水煤气变换反应。众所周知,这个过程需要催化剂。在铁矿石还原过程中,所有产品(Fe3O4、FeO 和 Fe)都被考虑作为可能的催化剂。其中特别活跃的是固态铁。因此,当存在金属 Fe 时,应将含 H2 的 CO/CO2 混合物中铁矿石的还原过程理解为反应序列。子反应(i)还原发生在氧化铁表面,而子反应(ii),通过水-气反应再生氢气,发生在铁表面。
两个子反应的空间分离需要通过传输过程将它们连接起来,传输过程将由反应参与者之一以气体扩散或表面扩散的形式发生。最佳条件出现在Fe/Fe氧化物/气体三相界。
还原时肿胀
铁矿石或球团的表观体积通常在还原过程中增加。这称为肿胀。大致可以看出三种溶胀行为。这些被称为 (i) 正常膨胀,(ii) 灾难性膨胀,其中随着 Fe2O 转化为 Fe,体积突然膨胀,Fe 以丝状生长的形式出现,称为纤维状 Fe 的晶须线,以及(iii) 爆裂膨胀,一种含有少量碱的富铁材料的典型行为。后一种行为与灾难性膨胀不同(尽管同样严重),因为膨胀的主要部分发生在 Fe 作为反应产物出现之前。
可以说,块状矿石和烧结矿都不会发生异常膨胀或灾难性膨胀,而某些类型的球团会这样,并且由于异常膨胀的球团柔软、海绵状且易于崩解,因此会降低负载的渗透性,从而导致操作问题.
文献报道的不同 Fe 氧化物和 Fe 的比容为每克 Fe 0.272 cc Fe2O3(室温下)、每克 Fe 0.270 cc Fe3O4、每克 Fe 0.231 cc FeO(23.5 % O2) ,以及每克 Fe 0.128 cc 的 Fe。因此,预计在每个减少阶段,交易量都会减少。然而,Fe矿石膨胀的主要原因是六方赤铁矿转变为立方磁铁矿并产生晶格紊乱。晶格扰动导致孔隙形成,在赤铁矿向磁铁矿转变过程中,铁矿石的表观体积显着增加。
一般而言,在富含 CO 的气体还原过程中,膨胀比在富含 H2 的气体中大得多。这种行为的原因是在含 CO 气体混合物中的 C 沉积过程中发生金属粉尘。然而,当没有 C 沉积时,很难解释在 CO-CO2 气体混合物还原过程中可能发生的膨胀。伴随复位而出现的肿胀或收缩的原因和影响尚未解决。
通常在矿球团中有两种杂质。它们是(i)对溶胀有阻碍作用的杂质,和(ii)对溶胀有改善作用的杂质。第一个例子是二氧化硅 (SiO2),第二个例子是碱金属 (K2O, Na2O)。已经注意到,含有高达 5% SiO2 的试剂级 Fe2O3 颗粒在 CO-CO2 气体混合物中还原时不会膨胀,并且酸性颗粒中需要一定量的 SiO2 以保持强度并防止灾难性膨胀。在第二种情况下,可以看出在 0.1% 至 1% 范围内添加少量碱金属 Na2CO3 或 K2CO3 会导致其他正常矿石球团在 H2 或 CO 中的灾难性膨胀。随着粒料中碱度(CaO/SiO2)比的增加,碱的影响变得更加明显。加入细粒酸性脉石形成稳定的碱金属硅酸盐可以防止这种不利影响。
对于球团矿中杂质(例如石灰含量)的影响,存在一些相互矛盾的观察结果。在赤铁矿球团中添加少量 CaO(小于 0.1%)会在还原过程中引起相当大的膨胀,这表明 CaO 是灾难性膨胀的原因。另一方面,已注意到在赤铁矿球团中添加约 1% CaO 可抑制还原过程中的膨胀。观察到的 CaO 对溶胀效果的这些变化可能是由于铁矿石中是否存在其他杂质,例如碱金属。
C还原赤铁矿
赤铁矿与 C 之间的反应在金属化矿石球团的制备中具有重要意义。回转窑工艺的发展激发了许多新的兴趣,该工艺在直接还原铁 (DRI) 的生产中使用固体 C 作为还原剂。一般认为,C 还原氧化铁是通过气态中间体 CO 和 CO2 发生的,但在非常高的真空下,真正的固-固反应是主要机制。
在赤铁矿被 C 还原过程中发生的通过气态中间体的反应机理是通过反应 (i) C(s) + 0.5 O2 =CO(g), (ii) FexOy(s) + CO(g) =FexO (y-1) (s) + CO2(g),和 (iii) CO2(g) + C(s) =2CO(g)。
CO 的初始形成是整个反应速率中的一个重要步骤。截留空气中的 O2 与 Fe 氧化物解离释放的 O2 气体与 C 反应生成 CO(第一反应)。此外,在 C 和 Fe 氧化物颗粒之间的接触点发生真正的直接还原也可以形成一些 CO。由此产生的CO气体容易与赤铁矿颗粒反应(第二反应)。 Boudouard 或 CO2 气体和 C 颗粒之间的溶液损失反应使 CO 气体再生(第三反应),从而趋于恢复样品孔隙内所含气相的还原电位。 CO2 中某些类型的 C 的氧化在某些金属和金属化合物的存在下被催化。通过添加 Li2O(氧化锂)已经观察到该过程的速率提高,并且已经报道了通过添加 FeS(硫化亚铁)的抑制效果。已发现金属铁是石墨 (C) 气化的良好催化剂。由于混合物中存在这种不可预测的催化反应,通过数学模型推导出的描述反应总速率的方程价值有限,只能适用于那些没有催化反应的系统。
在中等高温(例如 1000 摄氏度)下,氧化铁反应的速率(在高于 570 摄氏度的温度下,顺序为 Fe2O3、Fe3O4、FeO、Fe)远大于 Boudouard 反应的速率。换句话说,根据布杜尔反应,整个过程受到 CO 气体可用性的限制。因此,在稳态下,该气相的组成与 FexOy/FexO(y-1) 的平衡气相组成紧密对应。
碳氢化合物还原铁氧化物
碳氢化合物可以两种方式用作生产 DRI 的还原剂。 These are (i) direct use of hydro-carbons or a mixture of gas containing hydro-carbons, and (ii) use of the reformed hydrocarbon products (CO, H2), by reforming within the reduction reactor (it has been found that auto-catalytic reforming of some hydro-carbons within the reducing furnace provided an access of macro and micro porosity which leads to more extensive reduction and also which leads to the deletion of the capital cost of gas reformer and processing.
There are a few studies using directly hydrocarbons or a mixture of gas containing hydrocarbons as reductant for direct reduction of iron ores. Two important points emerge from these studies. The first is that the rate of reduction with hydrocarbons is slow and the production of a high quality of DRI is troublesome and uneconomical. The second point is that these studies have been done under isothermal conditions in a thermo-gravimeter with single particle or powder compact, thus the results are of only theoretical value.
Theoretical importance of investigations with hydrocarbons – The kinetics of ferric oxide reduction by pure methane (CH4) has been studied in the three temperature ranges of (i) low temperature (500 deg C to 600 deg C), (ii) medium temperature (650 deg C to 750 deg C) and (iii) high temperature (800 deg C to 950 deg C). At the low temperature, the reduction proceeds only from Fe2O3 to Fe3O4. A prolonged holding of the sample in a stream of CH4 has not led to any process extension beyond this stage. The rate became appreciable at 650 deg C. In special experiments after the Fe3O4 composition has been reached, the sample has been reduced further by H2 and CH4. It has been shown that CH4 reduction in the low temperature range beyond the Fe3O4 stage occurs only if a sufficient quantity of metallic Fe has been built up. In this case the reducing agent has not been CH4, but its decomposition product, H2. C formed by CH4 decomposition takes almost no part in the reduction and gets accumulated in the sample.
In the medium temperature range the conversion of Fe3O4 to FeO takes place but at low rates. A sharp rise in reduction rate is observed on going from 750 deg C to 800 deg C. The process becomes very sensitive to temperature changes beyond 800 deg C, and accelerated considerably in the high temperature range, when metallic Fe appeared in the sample. The appearance of metallic Fe at the FeO to Fe stage, at comparatively high temperatures indicates a decisive role of metallic Fe as a catalyst for reforming CH4 by the reduction products (CO2, and H2O). In the absence of a catalyst, the decomposition of CH4 and its reforming by the reduction products (CO2, H2O) do not occur to any substantial extent and no C accumulation in the sample has been observed. When the Fe catalyst is present, CH4 dissociation into the elements takes place only at very late stages of reduction, when there is insufficient CO2 and water vapour to convert all the CH4 diffused into the sample. C build-up in the sample starts from that stage.
In the 2-stage production of DRI with CH4, it has been found that the complete decomposition of CH4 in the presence of the Fe bearing material occurs at temperatures of 850 deg C to 900 deg C, which is 400 deg C to 450 deg C lower than on an inert surface (e.g. fire clay), while the reaction rate, conversely, has been 10 times higher. The products of the first stage are a sooty Fe containing 30 % to 50 % C and technically pure H2.
In the second stage, the product of the first stage (sooty Fe with highly dispersed C in the pores of DRI and on the surface of the Fe particles) has been used as an active reducing agent and mixed with mill scale or concentrate. The mixture has been reduced in the temperature range 1050 deg C to 1100 deg C with a make-up reducing agent of H2 reformed natural gas. The results of industrial trials has shown that the use of sooty Fe instead of soot, petroleum coke and the other known carbonaceous reducing agents considerably intensified the Fe-oxide reduction process. As is well known, the direct reduction of Fe oxides with C is directly related to the rate of reaction between the C and CO2. The sooty Fe can have intensified the rate of Boudouard reaction.
The isothermal reduction of hematite ore pellets (with 10 % to 15 % porosity) in a thermo-balance with a mixture of CH4-H2 (containing 4.5 % CH4) within the temperature range 700 deg C to 1000 deg C has shown that the reduction is chemical – controlled initially and diffusion – controlled in the later stages. It has been shown that reduction in pure H2 is faster than in the CH4- H2 mixture. This difference is attributed to C deposition in the outer reduced layers of the pellet, causing resistance to gas diffusion when the reducing gas contained CH4. It has been shown that the excess residual C can be removed from the reduced iron at lower temperature by its hydrogenation.
In another study, it has also been demonstrated that it is possible to hydrogenate residual C in direct reduced products to CH4. The C formed as a result of the reduction of Fe oxide in a mixture of CH4 and H2 (containing 20 % CH4) reacted with steam (H2O) according to the water gas reaction to regenerate H2 and produce CO.
Pure ferric oxide briquettes were reduced at temperatures ranging from 800 deg C to 1050 deg C, in gas mixtures containing H2, CO, CH4, N2 and CO2, which has been obtained by partial oxidation of natural gas with air. The CH4 content of the reformed gas mixture was between 13 % and 16 %. The overall reduction rate again has been controlled initially by chemical reaction and the gaseous diffusion has been applicable during the latter stages. It has been shown that the hematite ore briquettes have swelled and considerable porosity has been was developed during reduction. The solid-state diffusion rates increased more rapidly with temperature than it did by interfacial or gaseous diffusion reaction rates. The reduction of porous (30 % porosity) Fe ore in CH4 has indicated that the reaction proceeded stepwise from Fe2O3 to Fe3O4, FeO and Fe. The Fe catalyzed the CH4 cracking reaction. Optimum conditions for CH4 utilization occurred at around 1000 deg C.
The above findings are not consistent with the earlier studies on the understanding of high-grade porous (around 30 %) or dense hematite ore reduction kinetics, which had shown that the rate of reduction can be considered to fall between 3 limiting cases, namely (i) uniform internal reduction, (ii) limiting mixed control, and (iii) diffusion in porous iron layer, respectively with the rate of reduction corresponding to, (i) chemical control, (ii) the overall chemical control and diffusion control, and (iii) diffusion control. The overall rate of reduction is not controlled by only one of these rate controlling mechanisms and can be changed from one limiting case to another during the course of reduction.
In one of the studies it has been found that the most important factors controlling the extent of reduction are (i) the temperature, (ii) the composition of gas, presence of unreacted hydrocarbons in the reducing gas, the ratio of H2/C in it, and reducing capacity, (iii) the ore particle size, and (iv) the residence time for reduction.
Reduction of Fe oxides with the products of CH4 reformed with H, O within the reduction furnace – In early 1981 a commercial process has been introduced, using gaseous mixtures containing upto around 30 % by volume of CH4 (e.g. coke oven gas), for the direct gaseous reduction of Fe ore in a counter current moving bed shaft furnace. The furnace contained a reduction zone, a cooling zone, and an intermediate reforming zone. A hot mixture of coke oven gas and steam has been fed to the intermediate zone and reduced Fe ore therein catalyzed the reforming of the CH4 to CO and H2. The reformed gas flows upward into the reduction zone for the reduction of Fe ore.
制造工艺