

在供体-π-受体交替共轭聚合物中鉴定桥特异性分子内激子解离途径
摘要
分子内激子解离对于有机太阳能电池中的高效移动电荷载流子产生至关重要。然而,尽管备受关注,π桥对供体-π-受体(D-π-A)交替共轭聚合物中激子解离动力学的影响仍不清楚。在这里,使用飞秒时间分辨瞬态吸收 (TA) 光谱和稳态光谱的组合,我们跟踪了由秦的团队合成并命名为 HSD-A 的三种 D-π-A 交替共轭聚合物中的超快分子内激子弛豫动力学、HSD-B、HSD-C。发现加入噻吩单元作为π桥会导致稳态吸收光谱的红移。重要的是,我们揭示了由桥特异性电荷转移 (CT') 状态介导的新分子内激子解离途径的存在,在 π 桥接 HSD-B 和 HSD-C 中,TA 指纹峰位于 1200 nm。与 HSD-A 相比,这种 CT' 状态导致 HSD-B 和 HSD-C 的电子捕获率更高。取决于 CT' 状态的比例和非双生复合是理解 HSD-B 中比 HSD-C 中的高功率转换效率的重要步骤。我们认为这种桥特异性激子解离途径在有机光伏材料D-π-A交替共轭聚合物的超快分子内激子解离中起着重要作用。
<图片>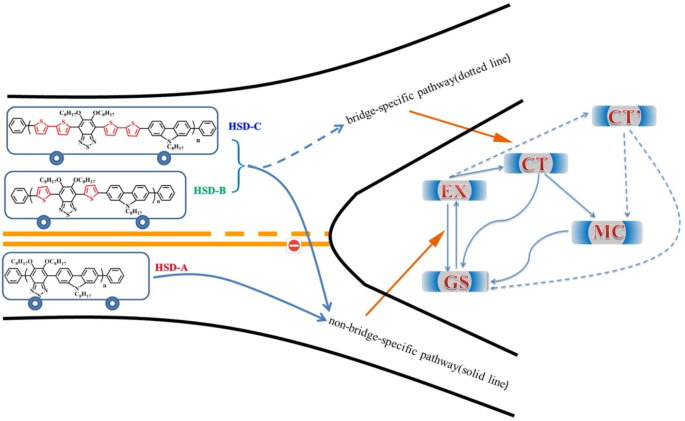
介绍
利用太阳能来满足世界不断增长的能源需求的有机光伏 (OPV) 设备可被视为清洁和可再生能源生产的最重要替代品之一 [1,2,3,4]。供体-受体交替共轭聚合物,其中不同电子亲和力的共轭嵌段沿着聚合物的主链交替排列,是优良的有机电子材料。它显示出相对较高的功率转换效率 (PCE) 的部分原因是光学间隙较低,可以更有效地收集近红外 (NIR) 范围内的太阳光子 [5]。因此,由供体-受体交替共轭聚合物组成的OPV器件可能成为硅基太阳能电池的经济可行的替代品[6,7,8,9]。
来自各个团体的最新研究表明,可以通过使用低带隙共轭交替共聚物(如 PCDTBT、PBDTTT 和 PTB 家族中所见)来提高 OPV 的效率 [10,11,12,13]。这些聚合物之间的一个重要区别是最低能级的激子跃迁表现出部分电荷转移特性。分子内电荷转移状态被认为促进异质结处的最终电荷分离 [14,15,16,17,18,19]。因此可以合理预期低带隙供体聚合物的性能与器件的性能密切相关。然而,器件性能与聚合物固有特性之间的联系尚不清楚。特别是,低带隙供体聚合物的超快激子分裂和载流子动力学与器件的 PCE 没有直接关系。一方面,超快和慢速时间尺度上的大量并行和顺序过程仅在与设备相关的条件下才能发现。另一方面,只有在供体 - 受体体异质结(BHJ)界面中的激子分裂才能被认为是调节器件 PCE [5]。因此,有必要研究低带隙给体聚合物的激子和载流子动力学,以优化D-A有机光伏器件的PCE。
最近,Qin 的团队合成了一系列 D-π-A 交替共轭聚合物 [20,21,22]。例如,HSD 共聚物由 2,7-连接的咔唑作为供体单元和 5,6-双(辛氧基)苯并[c][1,2,5] 噻二唑作为受体单元组成,同时具有不同的 π 桥。供体嵌段和受体嵌段直接聚合制备光伏材料HSD-A。不同的是,一个噻吩单元作为π桥连接供体块和受体块表示为HSD-B,供体块和受体块由两个噻吩单元连接表示为HSD-C。他们发现共聚物中的 π 桥对 HSD 共聚物的性能有显着影响。不同的 π 桥严重影响共轭聚合物主链的电子离域、薄膜的形态以及 HSD 共聚物的光学、电化学、电荷传输和光伏特性 [23]。使用HSD共聚物作为电子供体和PC71BM作为电子受体制备有机光伏器件,发现用具有不同π桥的HSD聚合物制备的器件具有不同的PCE。以 HSD-A:PC71BM 作为有源层的 OPV 器件表明 PCE 较低; HSD-B:PC71BM 的 PCE 为 5.4%; HSD-C:PC71BM 的 PCE 为 2.15% [20, 21]。这些证据表明,供体共聚物对聚合物太阳能电池的 PCE 有影响,但器件性能与供体共聚物的固有特性(如结构、能量和移动载流子动力学)之间的相关性尚不清楚。光吸收后的主要弛豫过程对于确定光伏器件的性能很重要。因此,通过跟踪激子来了解HSD共聚物的激子和载流子动力学势在必行。
超短激光技术的快速发展使得以飞秒时间分辨率和高空间精度监测和跟踪分子内化学键的形成和断裂以及分子内和分子间的各种动态过程成为可能。这项工作使用稳态吸收和飞秒时间分辨瞬态吸收光谱的组合阐明了 HSD 共聚物的激子解离和超快弛豫过程。详细测量和分析了特征谱带,揭示了激子解离动力学的超快弛豫机制。我们的研究结果更好地了解了HSD共聚物的物理性质,为提高聚合物太阳能电池的PCE提供了实验依据。
材料和实验方法
材料
HSD-A、HSD-B、HSD-C由Qin课题组提供,这些共聚低聚物的合成和表征见文献[20, 21]。这些共聚低聚物的分子结构如图 1a 所示。用于制备这些共聚低聚物的溶液是邻二氯苯,浓度约为 0.1 mg/ml。该浓度不仅可以保证可以测量出良好的时间分辨信号,而且可以保证发色团完全分离,从而在所用的激发强度下激发态不会被猝灭[24]。
<图片>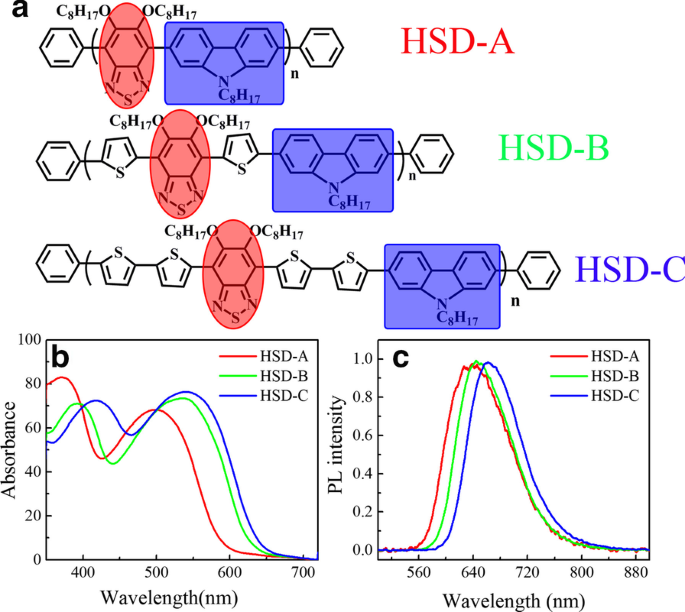
三种聚合物的分子结构(a ) 在这项工作中。蓝色阴影和红色阴影分别表示供体和受体部分。稳态吸收光谱 (b ) 和稳态光致发光光谱 (c )三个样品,HSD-A(红色),HSD-B(绿色),HSD-C(蓝色),在邻二氯苯中测量
光谱测量
稳态吸收光谱采用双光束分光光度计(Cary-5000,Agilent)测量,稳态荧光光谱采用光纤光谱仪(USB-4000,Ocean Optics)测量。
飞秒时间分辨瞬态吸收光谱由飞秒激光(相干)、光学参量放大器(OPA、TOPAS)和瞬态吸收光谱仪(Helios fire)测量。飞秒激光产生的飞秒激光通过分束器(1:1)分成两路,其中一路进入TOPAS,产生不同波长的泵浦脉冲;另一束光束再次通过分束器 (2:98),投影激光的一小部分进入 Helios 瞬态吸收光谱仪,以产生白光连续谱 (WLC) 探测脉冲(420-780 纳米、820-1600 纳米) ).
结果与讨论
图 1a 显示了本工作中使用的 HSD 共轭聚合物的结构式,供体部分用蓝色框标记,受体部分用红色圆圈突出显示。噻吩单元充当供体和受体之间的桥梁特异性,依次避免单个供体和受体单元之间的空间排斥。如前所述,它还可以实现供体和受体之间的长距离电荷分离,从而确保长寿命的电荷转移状态[8]。图 1b 显示了三种聚合物的稳态吸收光谱,具有不同 π 桥的三种聚合物的吸收光谱呈现相似的形状,具有两个不同的吸收带。在其他共轭聚合物中也报道了典型的双峰分布,这是 D-A 共轭聚合物的独特特征 [25,26,27,28,29,30]。 HSD-A 的吸收峰在 370 和 490 nm 附近,HSD-B 的吸收峰在 390 和 530 nm 附近,HSD-C 的吸收峰在 420 和 540 nm 附近。这两个吸收峰归因于与链内电荷转移相关的低能量峰的 π-π* 跃迁 [31]。稳态吸收峰的位置受不同 π 桥单元替换的影响,导致吸收峰红移主要是由于电子离域效应 [32]。我们已经对聚合物进行了量子化学计算,HSD 聚合物的前沿分子轨道已经计算并提供在图 2a 中。三个样品的最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)如图 2a 所示,HOMO-LUMO 能隙(ΔEH-L)绘制在图 2b 中。从图 2b 可以清楚地看出,从 HSD-A 到 HSD-B 和 HSD-C 的 HOMO-LUMO 能隙(ΔEH-L)逐渐减小,这与来自 HSD-A 的光谱的红移一致HSD-B 和 HSD-C [33]。图 1c 显示了三个样品在其溶液中的光致发光 (PL) 光谱。三个样品的 PL 光谱是相似的,并且与稳态吸收光谱一致。值得注意的是,随着噻吩数的增加,它们的峰向长波移动。 HSD聚合物的π桥可以调节由HSD聚合物和PC71BM共混制备的有机太阳能电池的PCE,我们在本研究中使用的器件的PCE按以下顺序列出:HSD-B> HSD-C> HSD- A [20, 21]。
<图片>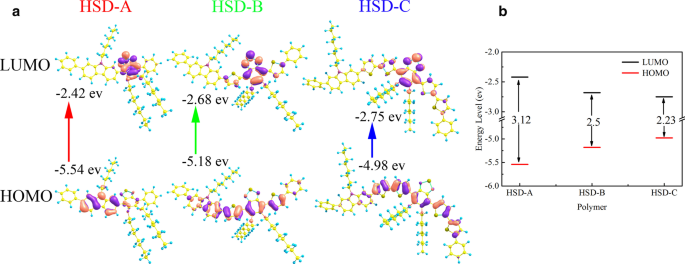
HSD 共聚物的前沿分子轨道 (a ) 使用 B3LYP-D3 泛函与 6-311G** 基组以及 HOMO 和 LUMO 能级 (b )
稳态光谱只能给出整体电子过渡态的宏观描述。为了研究 π 桥如何影响器件的 PCE,我们进一步对三种 HSD 聚合物进行了瞬态吸收测量,如图 3 所示。三个样品的可见光 (VIS) 范围图(图 3a– c) 相似,显示三个光谱特征。大约 500 nm 处的负信号(图中的浅蓝色)被指定为基态漂白 (GSB) 信号,因为它与图 1b 中所示的第二个稳定吸收峰很好地对应。所有三个样品在可见光范围内都有两个正吸收信号(图中的浅红色),吸收峰分别位于 600 nm 和 750 nm,这被认为是激发态吸收 (ESA) [34]。在近红外 (NIR) 检测范围内(图 3d-f),三个样品显示出明显的差异。 HSD-A在近红外范围内几乎没有吸收信号,而HSD-B和HSD-C在800-1500 nm范围内有大面积红色吸收信号。
<图片>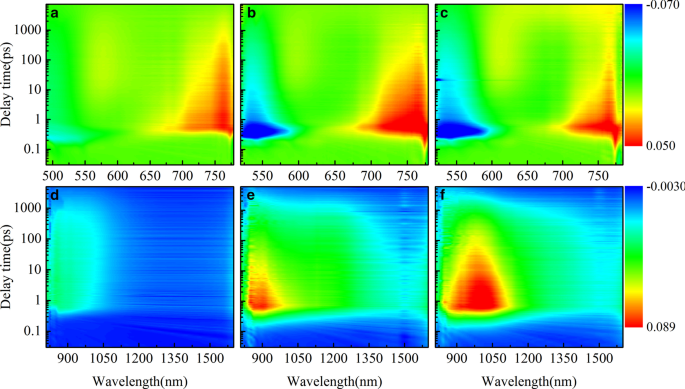
不同样品在不同探测波长下的飞秒时间分辨瞬态吸收 (TA) 光谱。在 HSD-A (a) 的 470 nm 波长处激发时,作为探测波长 (500–1600 nm) 函数的二维图 , d );在 500 nm 波长处激发 HSD-B (b , e );在 500 nm 波长处激发 HSD-C (c , f )
图 4 展示了三种 HSD 聚合物在超快时间尺度上的差分吸收光谱的时间演变。在 490-780 nm 范围内,在低光子能量下激发会在 700-780 nm 光谱区域产生广泛的正信号,该信号随着 490-600 nm 光谱区域的负信号迅速上升,作为 GSB。我们将 700 到 780 nm 的广泛正信号分配给激子 (EX) 态吸收,其归因如下。首先,它的寿命与文献中孤立聚合物中其他激子的寿命一致,为 500-1000 ps [35],远短于电荷转移 (CT) 状态和电荷分离 (CS) 状态的寿命,其时间尺度大于几纳秒。其次,它在最初的几百皮秒内具有与 GSB 类似的动态趋势。在几皮秒后出现另一个正信号(在大约 600 nm 处达到峰值),这对应于移动载波 (MC) 状态的形成。由于大约 600 nm 的光谱可以合理地归结为瞬态吸收实验中 GSB 和 MC 吸收的叠加这一事实。 600 nm 处的初始负信号是由于 GSB 的信号比 MC 吸收的信号强得多。随着延迟时间的增加,当MC吸收强于GSB时出现正TA特征。此外,在 650 nm 下沉的原因是由于受激发射 (SE) 以及与稳态荧光的光谱一致,不稳定的激发态返回到基态。在 NIR 范围内,可以看出三个样品的吸收信号在 1 ps 内增加,峰值在 1 ps 左右,然后呈现衰减趋势。有趣的是,三个样品的吸收信号的形状和衰减趋势是不同的。为了更详细地分析这些差异,我们对红外光谱进行了峰拟合,结果如图 5 所示。
<图片>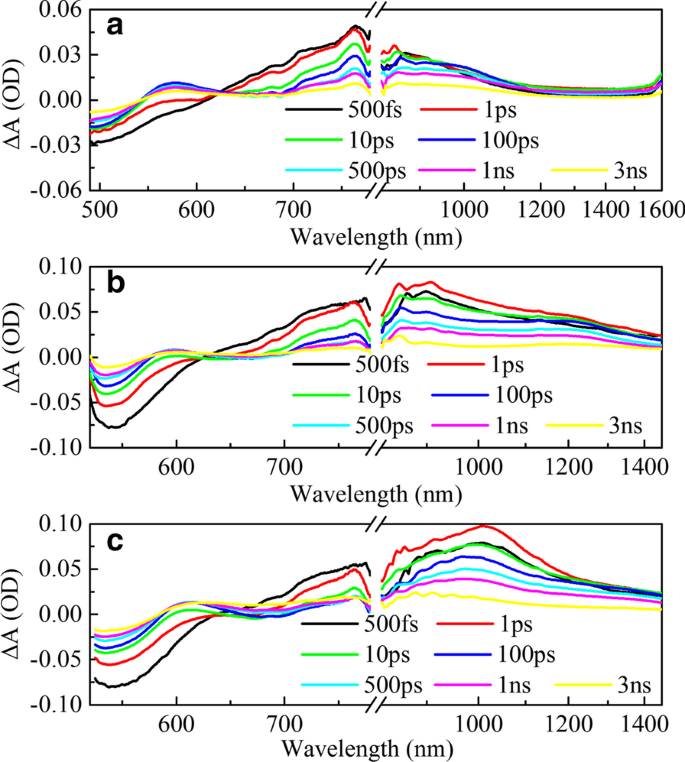
HSD-A (a) VIS-NIR 中的进化相关差异光谱 (EADS) ), HSD-B (b ), HSD-C (c )
<图片>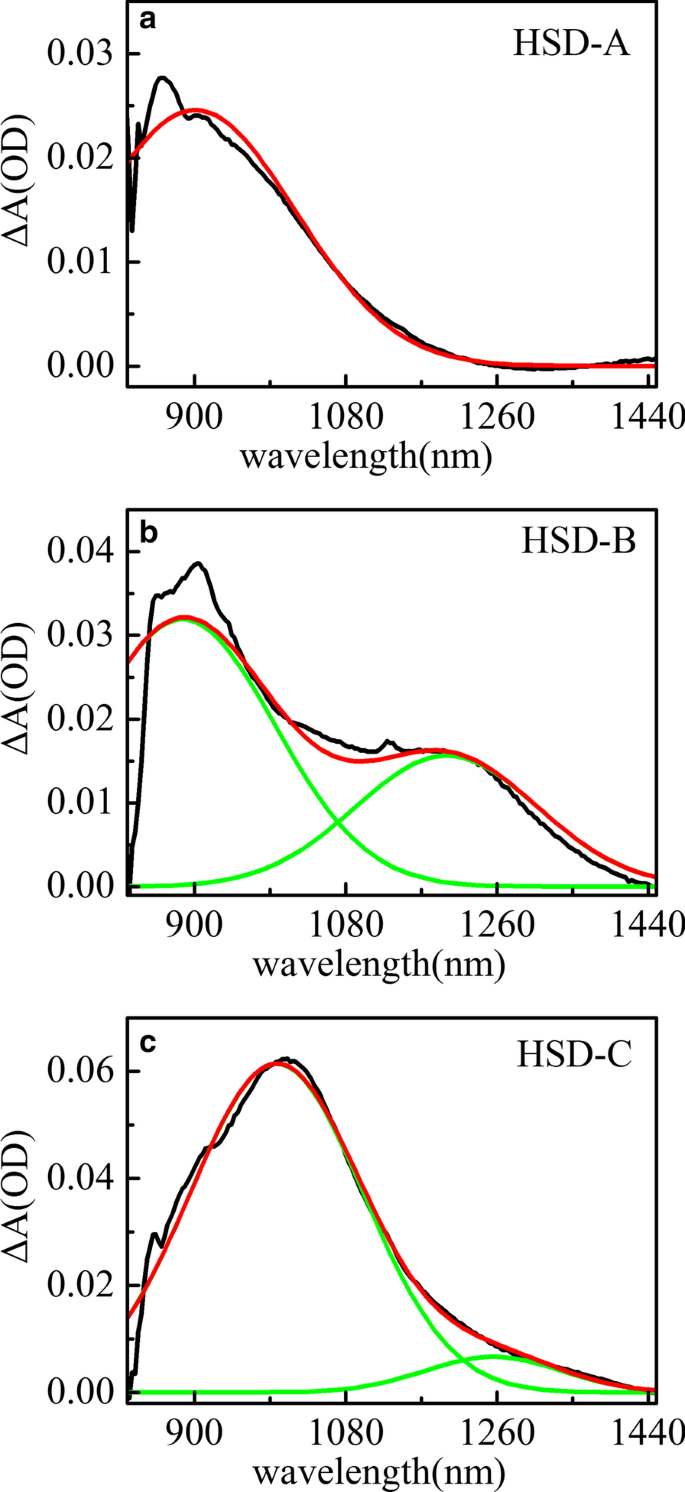
HSD-A (a ) 和 HSD-B (b ) 和 HSD-C (c ) 在红外范围内的延迟时间为 2 ps。黑色曲线代表样品在2 ps处的吸收光谱,红色曲线是拟合的吸收光谱,绿色是光谱中识别的光谱信号
图 5 显示了 HSD-A (a) 和 HSD-B (b) 以及 HSD-C (c) 在红外范围内延迟时间为 2 ps 的瞬态吸收光谱峰拟合。在 HSD-A 中,光谱可以通过一种成分分析很好地拟合,而在 HSD-B 和 HSD-C 中,这些光谱可以通过两种不同的成分分析来最大程度地近似。这意味着 HSD-B 和 HSD-C 在红外范围内比 HSD-A 剂量多一个吸收信号,这是由于添加了噻吩桥。噻吩桥的加入扩大了聚合物在 NIR 中的吸收范围,使 HSD-B 和 HSD-C 在 1200 nm 附近有一个新的吸收峰。在早期,这两个正信号的上升时间与 EX 峰值的衰减密切相关,如图 6 所示,这表明这些正信号是由 EX 状态直接产生的。所有三个样品中 900 nm 附近的正信号可以分配到分子内电荷转移 (CT) 状态。在这种状态下,激子分裂成空穴-电子对,空穴-电子对仍然足够接近以产生库仑引力[36, 37]。另一个新的1200nm附近的正信号仅存在于HSD-B和HSD-C中,并且早期也伴随着EX的衰减,但在长时间窗口下与CT状态的衰减趋势不同。这是一个新的激子解离通道,我们将其视为 CT' 状态。因为它具有与CT状态相似的特性,但衰减动力学与CT状态不同。
<图片>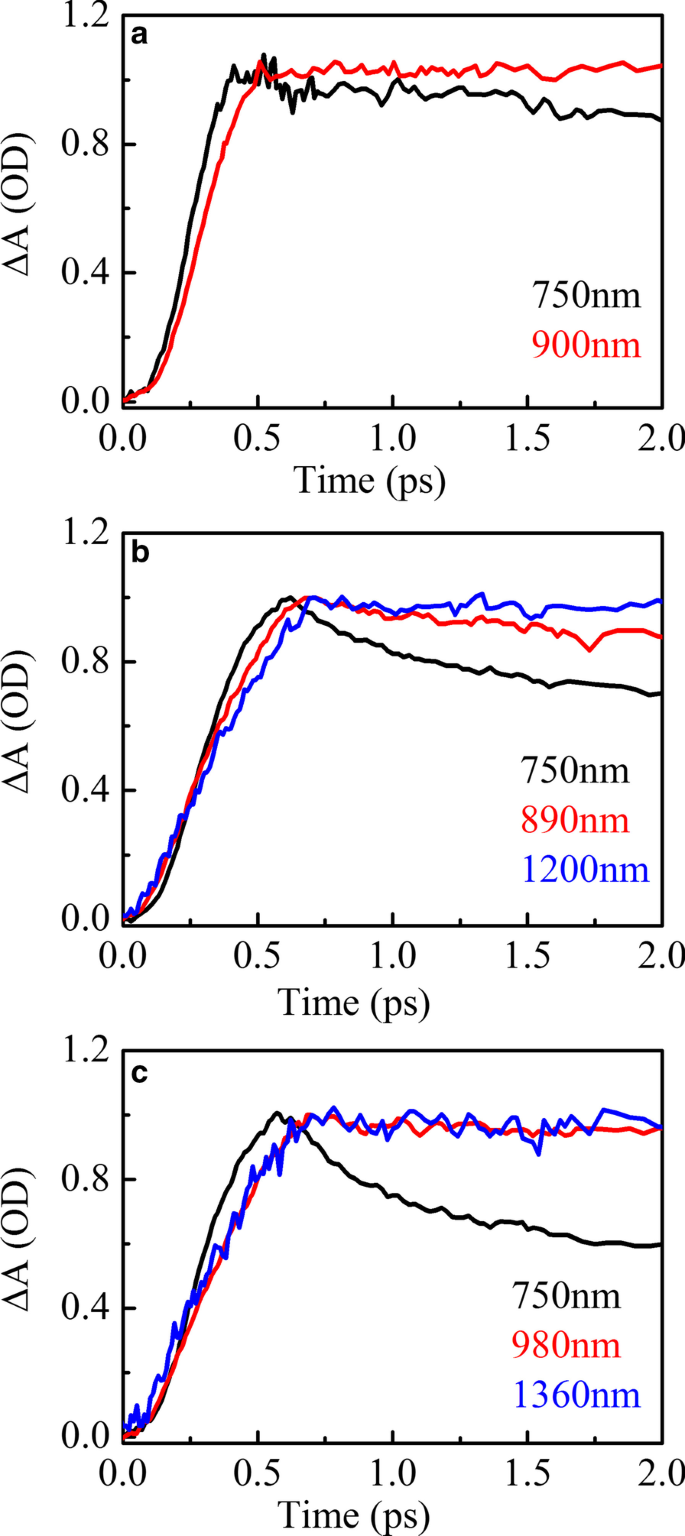
HSD-A (a) 的瞬态吸收 EX(黑色)和 CT(红色)和 CT'(蓝色)动力学 ) 和 HSD-B (b ) 和 HSD-C (c ),随着CT和CT'的增加同时表现出EX衰减
如图 7 所示,分别提取并拟合了 HSD-A、HSD-B 和 HSD-C 中 MC 状态、EX 状态、CT 状态和 CT' 的动态曲线,分别代表了不同组件。动态曲线的拟合公式为 ∆A(t) =a1exp(− t/τ1) + a2exp(− t/τ2) + ··· + anexp(− t/τn),其中 a1, a2, ...an 是幅度, τ1, τ2, …, τn 对应于时间常数 [38, 39]。表 1 列出了拟合的时间分量和相对幅度。为了比较,最大幅度被归一化。从MC状态拟合数据(图7a、b)可以清楚地看出HSD-A的载波生成速度是最快的。其形成寿命为 6.43 ps,比 HSD-B 的 12.6 ps 和 HSD-C 的 8.41 ps 短。然而,HSD-A 的载流子形成后衰减很快,而 HSD-B 和 HSD-C 有一个额外的缓慢上升过程,时间常数分别为 28.8 ps 和 26.4 ps。这可能会使 HSD-A 中的载波更难被捕获,这可能是设备 PCE 较低的原因之一。在 EX 状态下(图 7c、d),三个样品的衰减趋势明显不同。 HSD-A 的衰变寿命明显更长,因此激子分裂相对较慢。在 HSD-B 和 HSD-C 中,有 1 ns、1 飞秒(HSD-B 为 0.712 ps,HSD-C 为 0.408 ps)和一个皮秒(HSD-B 为 18.4 ps,HSD-B 为 7.96 ps)三个衰减寿命HSD-C) 生命周期代表 EX 状态到其他状态的转换。数百皮秒的更长寿命(HSD-B 为 735 ps,HSD-C 为 627 ps)与先前报道的孤立 P3HT 激子寿命处于同一数量级。因此,可以认为EX拟合中的\(\uptau _{3}^{{{\text{EX}}}}\)最有可能是没有跃迁过程的激子寿命[35, 40] .然而,在很长一段时间内,激子复合发生在 HSD-C 中。这三个样品的 CT 动力学(图 7e、f)最适合三个寿命组件。短暂的增加时间常数 \(\uptau _{1}^{{{\text{CT}}}} <1\) ps,与 EX 状态的并发衰减寿命密切相关,这意味着从EX 状态到 CT 状态。同样,CT态的衰变寿命\(\uptau _{2}^{{{{\text{CT}}}}\)与载流子态的增加寿命\(\uptau _ {2}^{{{\text{MC}}}}\),表示从 CT 状态到 MC 状态的转变。比较三个样品的CT状态衰减寿命,可以发现HSD-B的CT状态衰减寿命明显短于HSD-A和HSD-C,说明HSD-B的CT状态衰减速度更快速度。 HSD-B 和 HSD-C 的 CT' 状态(图 7g,h)在 50 ps 间隔内表现出与 CT 状态不同的动态曲线。它具有更长的过渡寿命\(\uptau _{2}^{{{\text{CT}}^{{\prime }} }}\),与\(\uptau _{3} ^{{{\text{MC}}}}\),表示从CT′状态到MC状态的转变。
<图片>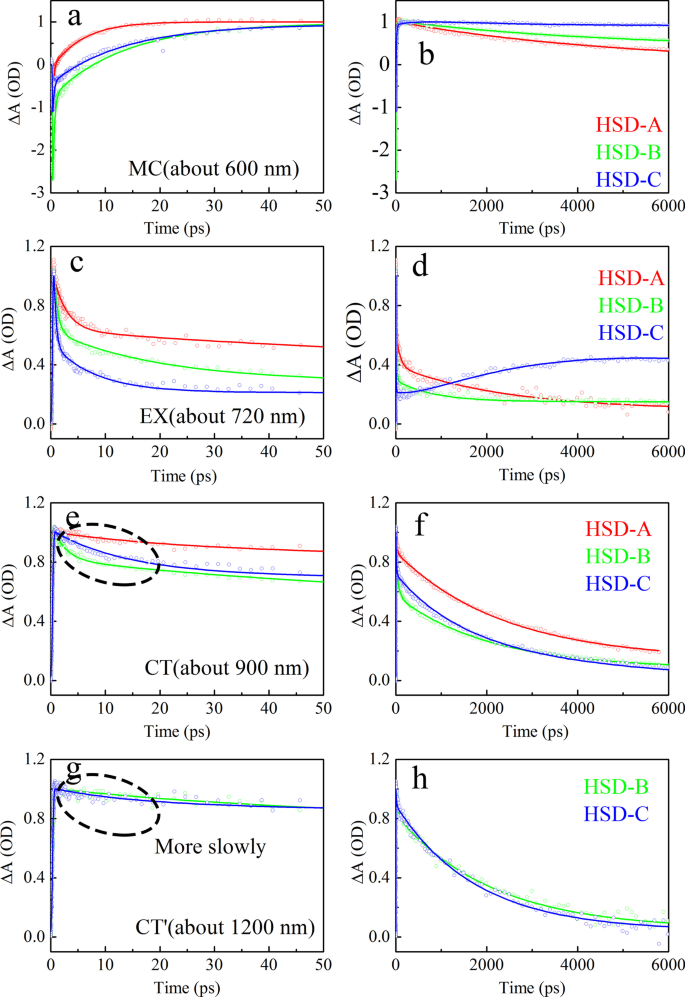
三个样品的所有瞬态光谱特性的动力学拟合。该图显示了 50 ps (a , c , e ) 和 5000 ps (b , d , f )。适合 MC (a , b ), EX (c , d ), CT (e , f ), CT′ (g , h ) 频谱特性
激子弛豫路径的简化能量图示意图如图 8 所示。聚合物中局部构象的差异导致不同状态的能量变化。因此,这些状态表现出不同的TA特性。在HSD聚合物中,不可避免地提出了MC态、EX态、CT态和CT'态来对激子弛豫机制给出更合理的解释。光激发后产生的激子会迅速分裂为CT态和CT'态。在这些状态下,激子分裂成空穴-电子对,并且仍然足够接近以体验库仑引力。随着时间的延迟,CT态和CT'态的电子-空穴将继续分裂成更稳定的MC态。重要的是,我们发现 CT' 状态仅存在于带有噻吩桥的 HSD-B 和 HSD-C 中,这为 HSD 聚合物增加了一个新的激子分裂通道。这将导致 HSD-B 和 HSD-C 的电子捕获率更高,这与与 HSD-A 相比,HSD-B 和 HSD-C 的 PCE 更高。同时,HSD-C 的 PCE 低于 HSD-B 的事实可以合理地解释如下:(a)添加两个噻吩作为 π 桥可能会增加 D 和 A 之间的空间分离,导致非双生HSD-C 中的重组。 (b) 从图 5 可以看出,HSD-B 的 CT' 状态比例明显高于 HSD-C,这决定了 HSD-B 的整体 PCE。因此,HSD聚合物中的CT'态对于获得更高的电子捕获能力至关重要,从而导致HSD聚合物器件具有更高的PCE。
<图片>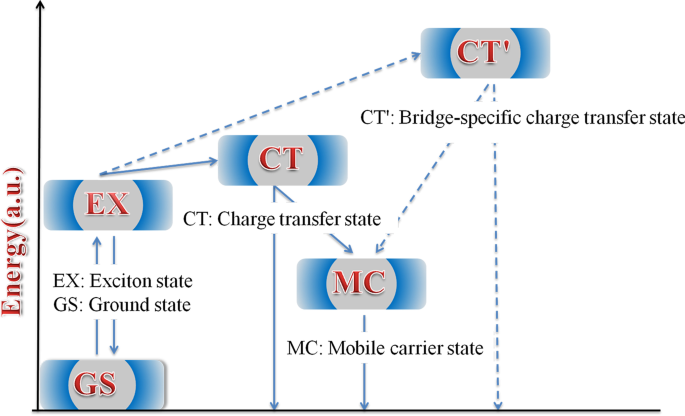
溶液中HSD聚合物激子分裂动力学状态和途径示意图
结论
总的来说,我们结合使用稳态和瞬态吸收光谱来研究 π 桥对 HSD 共聚物的影响。发现加入噻吩单元作为π桥会导致稳态吸收光谱的红移。同时,瞬态吸收数据表明以噻吩单元作为 π 桥的 HSD-B 和 HSD-C 具有额外的 CT' 状态,TA 指纹峰位于 1200 nm,这为 HSD 聚合物增加了一个新的激子解离通道。 CT'态的存在使得聚合物有利于光电转换。在这项工作中研究的三种 HSD 聚合物中,只有包含 π 桥的 HSD-B 和 HSD-C 具有 CT' 状态。因此,我们认为π桥的存在促进了CT'态的产生。但是,π 桥不能太长。例如,具有两个噻吩作为 π 桥的 HSD-C 聚合物由于存在过长的 π 桥而导致非偶合复合,并影响 CT' 状态的比率。此外,我们还通过分析所有瞬态光谱特性的动态拟合阐明了激子的弛豫途径。这些发现为提高共轭聚合物的能量转换效率和有机太阳能电池的进一步发展提供了重要的光物理信息。
数据和材料的可用性
本研究中使用和分析的数据集可根据合理要求向相应作者索取。
缩写
- D-π-A:
-
供体-π-受体
- 助教:
-
瞬态吸收
- CT′:
-
桥特定电荷转移状态
- OPV:
-
有机光伏
- PCE:
-
电源转换效率
- 近红外:
-
近红外
- BHJ:
-
体异质结
- TOPAS:
-
光参量放大器
- 无线控制器:
-
白光连续体
- PL:
-
光致发光
- VIS:
-
可见
- GSB:
-
基态漂白
- 欧洲航天局:
-
激发态吸收
- EADS:
-
进化相关差异谱
- EX:
-
激子态
- CT:
-
电荷转移状态
- CS:
-
电荷分离状态
- MC:
-
移动运营商状态
- SE:
-
受激发射
纳米材料