

五硅石墨烯作为锂离子电池高性能负极材料的第一性原理研究
摘要
根据第一性原理计算,预测一种新型的五边形 Si/C 复杂性作为一种有前途的锂离子电池负极材料具有潜在的应用价值。发现五硅石墨烯(P-Si2C4)的结构和热稳定性优于仅由碳原子组成的五石墨烯。电子能带结构分析表明空C-2p z P-Si2C4 中的状态提供了空间来容纳和稳定来自锂的电子,这使得锂存储在能量上有利。因此,P-Si2C4的一个分子式单元可以存储四个锂原子,对应的理论重量锂存储容量为1028.7 mAhg -1 .吸附锂的P-Lix的金属电子结构 Si2C4 以及非常小的锂迁移能垒有利于电池的快速充电/放电性能。讨论了 P-Si2C4 上锂吸附相互作用的机制。这些结果证明了一种设计用于高性能锂离子电池的二维Si/C复合负极材料的新策略。
背景
目前商业化的锂离子电池(LIBs)相对较低的能量密度难以满足商用电动汽车(EVs)的需求,成为电动汽车行业发展的一大挑战[1, 2]。为了提高LIBs的能量密度,我们需要提高电极材料的容量。由于其非常好的循环性能,石墨是应用最广泛的负极材料,但其理论重量容量(372 mAhg −1 ) 相对较低 [3, 4]。另一方面,硅具有极高的理论重量容量,约为 4200 mAhg −1 [5],但由于其在完全锂化状态下的体积膨胀非常大,高达 420%,因此循环性能很差 [6]。为了充分利用硅负极和碳负极的优势,设计一种Si/C复合负极在学术和技术上都具有重要意义。
Siligraphene 是一种二维 (2D) 类石墨烯层状材料,其中 C 原子部分被 Si 原子取代,首先根据第一性原理计算预测为稳定的 2D 材料 [7,8,9,10] 并已被从实验中成功制备 [11, 12]。林等人。已经表明可以通过溶液剥离技术制备 2D SiC 片 [11]。他们还成功地制备了可以在空气中保存数月的准二维 SiC2 片 [12]。后来,第一性原理计算表明,硅石墨烯是一种很有前途的负极材料,其理论容量为 1520 mAhg -1 和 1286 mAhg −1 分别为 g-SiC5 和 g-SiC2 [13]。结果表明,硅石墨烯负极继承了石墨负极的高循环稳定性以及硅负极的高容量。预测的高锂存储容量归因于增强的锂吸附与硅石墨烯单层的相互作用,这与 sp 中 Si 原子的变化有关 2 sp 3 - 像[13]。然而,电子构型从 sp 2 sp 3 在硅石墨烯上吸附锂的过程中,-like 伴随着明显的结构变化。这对于作为锂离子电池负极材料的硅石墨烯的循环性能不利。更好的解决方案是设计已经具有 sp 的 Si/C 复合材料 3 - 类似电子配置。
碳有多种同素异形体,由sp构成 , sp 2 , 和 sp 3 杂交或它们的组合。非常稳定的sp 2 加上石墨碳中的大 π 键电子配置是导致石墨烯上弱锂吸附相互作用的原因。当锂吸附在单层石墨烯上时,电荷从锂转移到石墨烯层 [14]。然后,Li 带正电并通过有吸引力的库仑相互作用与石墨烯层结合。然而,石墨烯层上锂的过量电荷破坏了石墨烯的大π键,这在能量上是不利的。因此,单层石墨烯上的锂吸附不利于吸附能负高于体心面的内聚能(bcc ) 相锂金属,这在锂离子电池中是不允许的。因此,不允许使用原始石墨烯单层存储锂 [15]。或者,与石墨碳材料相比,硬碳材料提供更高的 Li/Na 存储重量容量 [16,17,18]。硬碳材料被称为无定形相,由 sp 和 2 和 sp 3 碳原子[19]。硬碳材料较高的锂储存重量容量是否可能与sp有关? 3 电子配置?
五石墨烯,被称为 sp 2 -sp 3 混合二维碳同素异形体 [20],从第一性原理计算 [21] 被预测为锂/钠离子电池的有前途的负极材料。作为一种二维碳同素异形体,与具有蜂窝结构的传统石墨烯相比,五石墨烯具有更强的锂吸附行为。这种不同的锂吸附行为是否也与sp有关? 3 - 像五石墨烯中的电子配置?如果答案是肯定的,其背后的内在机制是什么?
尽管五石墨烯被预测为动态稳定的碳同素异形体,但与全球最稳定的相(石墨或石墨烯)相比,其内聚能明显更高。五石墨烯的内聚能比单层六方石墨烯的每个原子高约 0.9 eV [20, 22],这使得工业上大规模制造五石墨烯变得非常困难(如果可能的话)。然而,对于作为负极材料的应用,大规模制造非常重要。请注意,在硅烯中发现屈曲,因此 Si 与 sp 更稳定 3 - 类似于sp的杂交 2 [23,24,25] 而 C 原子更喜欢 sp 2 二维结构中的杂交;推测替换 sp 是合理的 3 在五石墨烯的结构中具有Si原子的-like C原子将在能量上受到青睐。我们称这种结构为五硅石墨烯。最近的实验表明,可以在Ag(110)上生长五边形Si基纳米带[26],表明Si基五边形结构的形成在实验上是可能的。
从理论上讲,Lopez-Bezanilla 等人研究了五硅石墨烯(P-SiC2)的电子和键合性质。他们发现 P-SiC2 表现出 p-p-σ 和 p-p-π 电子能带垂直排序的部分反转 [27]。随后,研究了 P-SiC2 的电子传输特性,并将其与五-石墨烯和五-CN2 [28] 进行了比较。有趣的是,P-SiC2 的电子传输性能可以通过应变工程进行调节,并且预测单轴压缩应变能够将单层五-SiC2 的空穴迁移率提高到 1.14 × 10 6 厘米 2 V −1 s −1 [29]。尽管结构相似,但与五-石墨烯相比,五-硅石墨烯具有不同的传输特性。由胡等人发现。五-石墨烯的热导率通过拉伸表现出标准的单调降低,而五-碳化硅具有不寻常的非单调上下行为[30]。五硅石墨烯的这些有趣特性与结构中硅原子的电子和化学性质密切相关。还发现硅元素本身有利于增强锂的吸附,因为与石墨烯相比,硅烯上的锂吸附相互作用要强得多 [31, 32]。因此,五硅石墨烯是否可用作锂离子电池的负极材料可能会很有趣。
在这项工作中,我们用第一性原理计算研究了五硅石墨烯中锂离子的存储行为,并具体讨论了五硅石墨烯如何存储锂离子的机制。我们从五硅石墨烯的热力学稳定性开始我们的研究,然后详细分析了锂吸附在其上的内在相互作用。最后讨论了五硅石墨烯作为锂离子电池负极材料的性能。
计算方法
这项工作中的所有计算都是使用基于密度泛函理论 (DFT) 的 Vienna Ab initio Simulation Package (VASP) [33] 进行的。投影仪增强波 (PAW) 方法 [34, 35] 结合通用梯度近似 (GGA) 交换和由 Perdew-Burke-Ernzerhof (PBE) 参数化的相关函数 [36]。对于所有计算,平面波的截止能量选择为 450 eV。晶格参数和离子位置完全松弛,最终力收敛到 0.02 eV/Å。电子能带结构是用 Heyd-Scuseria-Erznerhof (HSE06) 混合泛函计算的 [37],因为混合泛函对电子结构有更准确的描述。态密度(DOS)的计算采用高斯拖尾方法拖尾,拖尾宽度为0.05 eV。 Monkhorst-Pack [38] k 使用点采样和 k 的密度 -mesh 比 0.05 Å −1 厚 用于从头分子动力学 (AIMD) 模拟和 0.03 Å −1 用于其他计算。原子电荷分布通过巴德电荷分析[39]进行分析。锂离子迁移路径使用爬升图像轻推弹力带(CINEB)方法进行优化[40]。吸附能E 广告计算方式为:
$$ {E}_{\mathrm{ad}}=\left({E}_{\mathrm{host}+n\mathrm{Li}}-{E}_{\mathrm{host}}-{nE }_{\mathrm{Li}}\right)/n $$其中 E 主机,E Li 和 E host + Li 分别是主体五硅石墨烯材料、锂原子和吸附锂的主体的总能量,n 表示吸附在五硅石墨烯上的锂离子的数量。使用带有贝克-琼森阻尼的 DFT-D3 方法测试范德华 (vdW) 相互作用对吸附能的影响 [41]。除了吸附能外,平均锂嵌入电位(vs Li + /Li) 可以直接从 V 的 Li 金属(bcc 相)的吸附能和内聚能的差异中获得 ave =− (E ad − E Li − cohesive),如果我们分别选择eV和V作为能量和势能的单位。
结果和讨论
五硅石墨烯的结构和稳定性
五石墨烯的结构(见图1a,在本文下面记为P-C6)具有P-421 m 对称性(空间群编号 113)。优化的晶格常数为a =b =3.636 Å,与之前的结果一致 [20, 21]。在结构中可以发现两种类型的碳原子,即4-配位碳(在图1a中表示为C1)和3-配位碳(在图1a中表示为C2)。从碳原子的局部几何结构,我们可以看出 C1 是 sp 3 -like 杂交而 C2 是 sp 2 - 像杂交的。虽然C2原子被认为是sp 2 - 类似杂化[20],双C2-C2键特征使C2原子的化学性质与石墨烯不同,这将在本文后面详细讨论。在P-C6结构中用Si原子替换C1原子,形成五硅石墨烯(见图1b,优化结构的结晶信息文件在附加文件1:补充材料的SI-1中给出)并表示在本文的下文中作为 P-Si2C4。由于Si原子的原子半径大于C原子的原子半径,P-Si2C4的晶格常数(a =b =4.405 Å) 大于 P-C6,同时与其他报道的结果 [27,28,29,30] 一致。
<图片>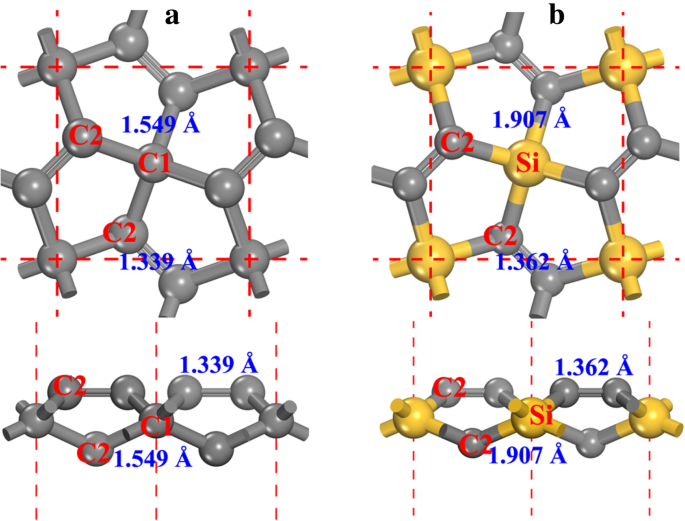
一 五石墨烯和b的球棒模型 五硅石墨烯。提供了俯视图(向上)和侧视图(向下)。灰色和黄色球体分别是 C 和 Si 原子。 4-和3-配位的碳原子分别表示为C1和C2。键长也显示在每个键旁边
为了评估相对热力学稳定性,表 1 列出了 C、Si 和 C/Si 配合物的不同同素异形体的内聚能。尽管从 ab initio 分子动力学 (AIMD) 模拟 [20] 表明五-石墨烯 (P-C6) 在 1000 K 下是稳定的,但 P-C6 的内聚能 (- 8.24 eV·atom -1 ) 远高于单层石墨烯 (− 9.14 eV·atom −1 )。这说明P-C6的量产一定非常困难。另一方面,P-Si2C4 的内聚能 (− 7.26 eV·atom −1 ) 仅比其最稳定的同素异形体 g-Si2C4 (− 7.46 eV·atom −1 ),表明与 P-C6 相比,五硅石墨烯的制备要容易得多。为了验证 P-Si2C4 的结构稳定性,计算了 P-Si2C4 的声子色散曲线并显示在图 2 中。尽管在 Γ 点附近的小区域(0.0039 THz 或 0.13 cm<)中发现了很小的虚频率sup> −1 ),我们仍然可以相信系统是动态稳定的,因为人们普遍认为这些小的虚频率(不大于 1 cm −1 ) 可能是模拟的产物 [42]。在其他动态稳定的 2D 材料中也报道了虚频率,例如锗烯 [43] 和砷烯 [44]。通过提高计算精度或采用不同的计算方法等技术处理,可以去除这些虚频。
<图片>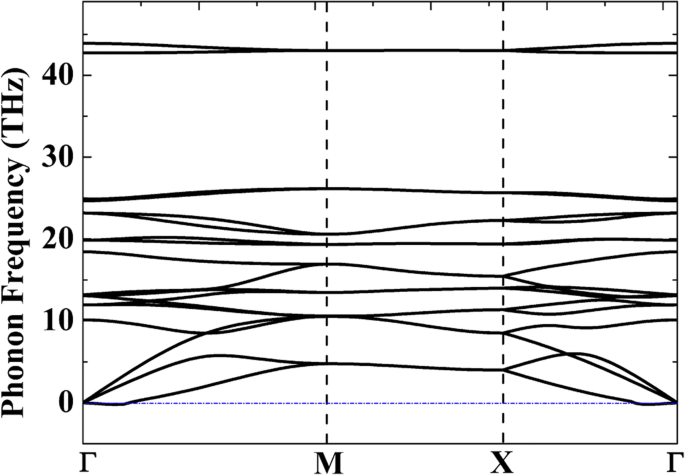
线性响应理论计算的二维P-Si2C4单层声子色散曲线
此外,还进行了 AIMD 模拟以评估 P-Si2C4 在高温下的结构稳定性。在 1000 K、1500 K、2000 K 和 2500 K 的温度下,使用 3 × 3 和 4 × 4 超胞在规范集合中执行 AIMD(参见附加文件 1:图 S1)。附加文件 1:图 S2 和 S3 分别显示了在不同温度下使用 3 × 3 和 4 × 4 超晶胞进行 AIMD 模拟结束时 P-Si2C4 的原子配置。如图所示,在 20 ps 的模拟时间内,当温度高达 2000 K 时,五边形原子环保持不变,表明该结构可以承受高达 2000 K 的温度。另一方面,严重的结构变形观察到六边形环(见附加文件 1:图 S2d)以及其他缺陷(附加文件 1:图 S3d)出现在快照中,表明结构在 2500 K 时被破坏。在 P-Si2C4 中发现的六边形环在 2500 K 下,g-Si2C4(由六边形环组成 [13])比五相 P-Si2C4 更稳定,与表 1 中给出的内聚能一致。这些结果证实了结构的稳定性P-Si2C4比P-C6稳定得多,只能承受1000 K的温度。
Li 在五硅石墨烯上的吸附
为了研究锂在五硅石墨烯 P-Si2C4 上的吸附,考虑了不同的锂吸附位点,松弛后可以找到四个稳定的吸附位点(如图 3a 所示)。稳定的Li吸附位点是Si原子的顶部位点(表示为T),Si2C3五边形环的空心位点(表示为H),以及下层(B1)和上层(B1)的两个C2原子之间的桥位( B2)。锂离子在这些位点上的吸附偏好可以通过表 2 中的吸附能来表征。结果表明,最稳定的锂吸附位点是 B1 位点,吸附能为 - 1.922 eV。另一方面,H 位点(- 1.905 eV)的吸附能与 B1 位点非常接近。 Li 吸附能也由吸附高度表示,因为较低的吸附能对应于较小的吸附高度(见表 2)。在锂吸附过程开始时,锂离子优先停留在最稳定的 B1 位点。在所有 B1 位点都被占据后(对应于 Li2Si2C4 的化学计量,见图 3b),锂离子开始停留在 H 位点。由于 B1 和 H 位点之间的距离非常小(~ 1.5 Å),B1 和 H 位点的锂离子发生强烈的排斥相互作用。结果,B1 位点的锂离子被排斥到附近的 H 位点,因此 B1 位点变空,而所有 H 位点都处于 Li4Si2C4 状态(见图 3c)。还测试了 vdW 相互作用对 Li 吸附能的影响,结果在表 2 的括号中给出。 如图所示,vdW 相互作用对不同吸附位点的吸附能的贡献从 - 0.12 到 - 0.17 eV,表明vdW相互作用有利于Li吸附。
<图片>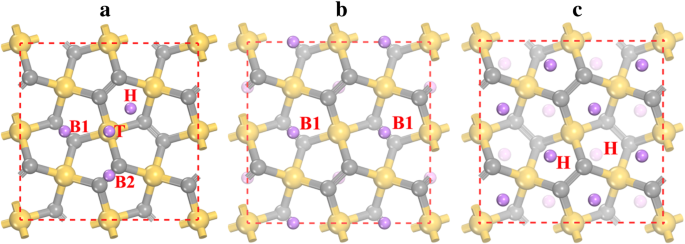
a上的锂吸附位点 五硅石墨烯表面和b最稳定结构的原子构型 Li2Si2C4 和 c Li4Si2C4。黄色(最大)、灰色(中等大小)和紫色(最小)球体分别是 Si、C 和 Li 原子。 H、T、B1 和 B2 表示锂吸附位点
锂吸附能与bcc的内聚能比较 锂金属相(− 1.86 eV·atom −1 ),我们可以判断锂吸附是否具有电化学活性。如果吸附能低于锂金属的内聚能,则有利于锂离子吸附,吸附对应于正放电电位。如表1所示,B1和H位的吸附能均低于- 1.86 eV,表明B1和H位都是锂存储的电化学活性位。为了评估锂存储容量,不同锂离子浓度下的锂吸附能x (Li/(Si + C) 比) 计算并与 bcc 的内聚能进行比较 利金属。如图4所示,当Li/C比x时,Li吸附能低于- 1.86 eV 小于 2/3,对应于 P-Si2C4 的一个晶胞中吸附了 4 个锂原子,理论重量容量为 1028.7 mAhg -1 (Li4Si2C4)。与锂存储容量相比,电池的能量密度等于容量乘以输出电压。好的阳极材料应该具有相对较低的电化学势,这可以从吸附能中获得。平均电位约为 0.1-0.2 V,相对较低,有利于全电池系统的更高输出电压。此外,图 4 还显示了不同 Li 吸附浓度下的晶格常数变化。如图所示,P-Si2C4 的晶格常数在锂吸附时略微收缩。当浓度x 为1/6,晶格变化达到最大值,小至- 0.94%,说明充放电过程中体积变化很小。较小的体积变化有利于在循环过程中保持P-Si2C4的结构稳定。
<图片>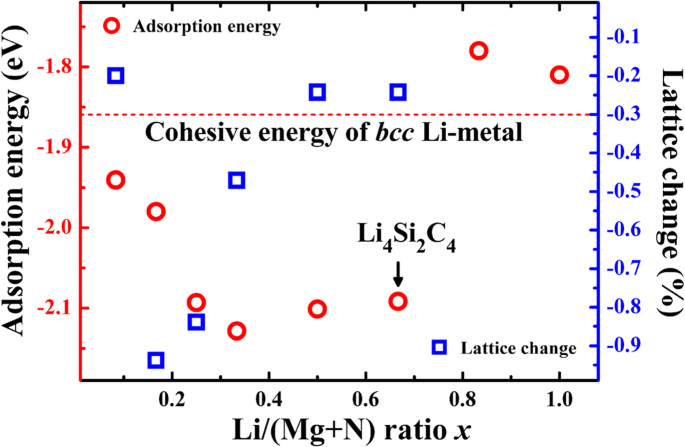
P-Si2C4 表面上计算出的锂吸附能(红色循环,左手轴)和晶格常数变化(蓝色方块,右手轴)作为锂吸附浓度(Li/(C+Si) 比)的函数)。 bcc的内聚能 锂金属相也包括在内用于比较
锂吸附后五硅石墨烯的电子结构分析
五硅石墨烯 (P-Si2C4) 的电子能带结构及其锂化状态如图 5 所示。 正如所见,P-Si2C4 是一种间接带隙约为 2.35 eV 的半导体,相比之下要小得多具有 3.46 eV 的五石墨烯 P-C6(参见附加文件 1:图 S4)。 P-Si2C4 比 P-C6 更小的带隙源于最高占据带(图 5a 中的第 11 和 12 号)的增强色散,特别是在高对称性 M 和 Γ 点。带隙在 12 号(最高占据带)和 13 号(最低未占据带)能带之间打开。 12号能带的能级在M点明显升高,费米能级升高,带隙减小。
<图片>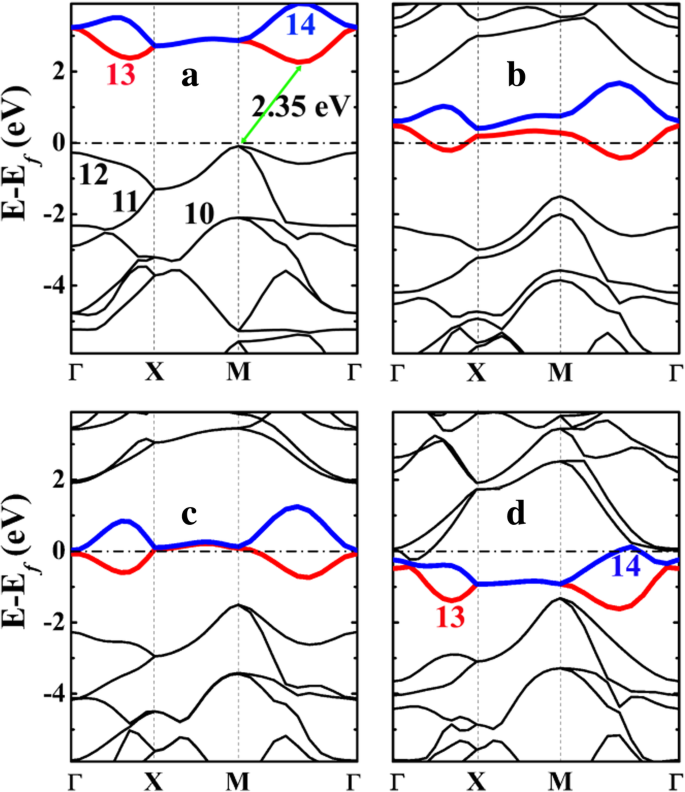
a的电子能带结构 五硅石墨烯P-Si2C4及其锂化状态b P-LiSi2C4,c P-Li2Si2C4 和 d P-Li4Si2C4 由 HSE06 计算。费米能级选择为 0 eV。 a 中从 10 到 14 的数字 和 d 表示乐队的编号,而乐队编号 13 和 14 分别用红色和蓝色突出显示。能带标记符合VASP代码,其中能带标记参考价带和导带,标记不包括核心电子
从投射到图 6 所示的 10-14 号带的电荷密度(波函数)的形状分析,我们可以看到,12 号带对应于 C 和 Si 之间形成的 σ 键的键合状态原子,而带号 13(和 14)对应于 2-p C 原子的 z 状态。空的 C-p z态为锂吸附提供了容纳和稳定电子的空间,这使得锂吸附过程在能量上有利。
<图片>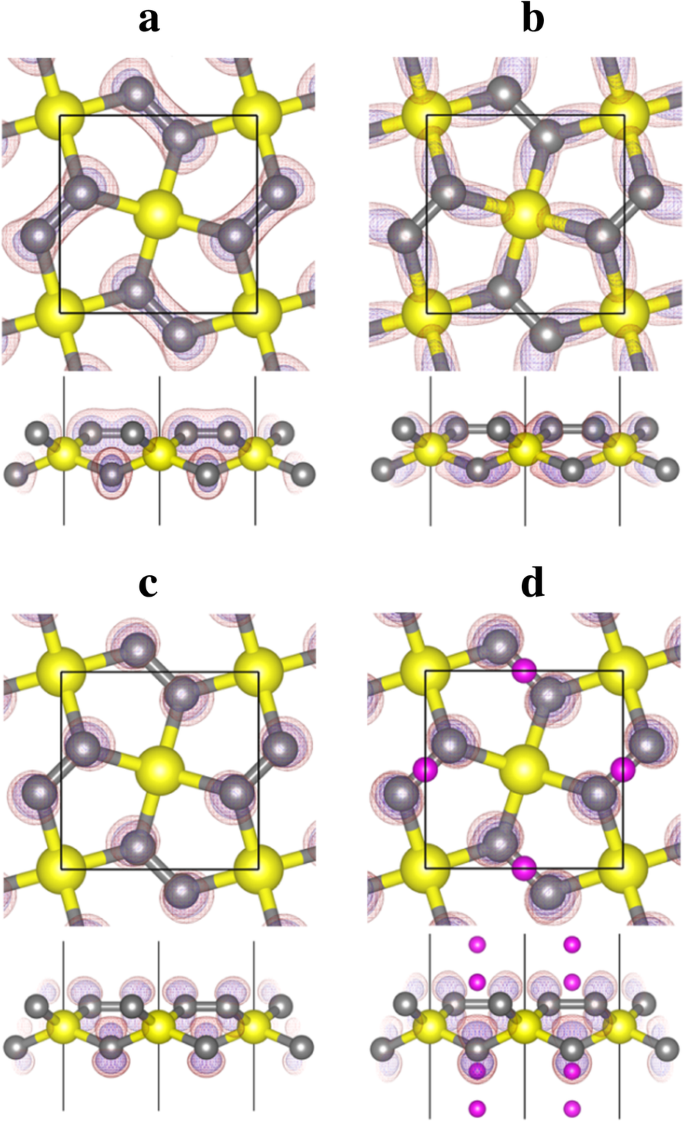
带 a 的带分解电荷密度等值线 第 10 名,b 第 12 号和 c 13号五硅石墨烯(P-Si2C4,图5a)和带d 第 13 号锂化五硅石墨烯(Li4Si2C4,图 5d)。黄色(大)、灰色(中型)和紫色(小)球体分别是 Si、C 和 Li 原子。电荷密度等值线以透明红色显示(等值面值为 0.02 e/Å 3 ) 和蓝色(等值面值为 0.01 e/Å 3 ) 颜色
P-Si2C4 中最高占据能带的增强分散可归因于两个因素:首先,电子占据带 12 号(C-Si σ 键)和带正电的 Si 原子之间的库仑吸引力,与在纯碳五石墨烯 P-C6 中。 Bader 电荷分析表明,在五硅石墨烯 P-Si2C4 中,Si 原子带正电。如表 3 所示,硅石墨烯或五硅石墨烯中 Si 原子的贝德电荷约为 1.65 e , 表明 Si 原子带正电 + 2.35 e .另一方面,五石墨烯 (P-C6) 中的 C1 原子也带正电,但净电荷仅为 + 0.08 e .因此,与 P-C6 相比,P-Si2C4 中除了共价键相互作用外,C 和 Si 原子之间发生了强库仑相互作用。这有利于费米能级附近的占据能带(第 12 号)的分散。其次,P-Si2C4 中增强的屈曲也可能有助于第 12 号带(C-Si σ 键的键合状态)的分散,因为更大的屈曲表示更多的 sp 3 -杂化和更强的σ键在C-Si原子之间形成。这也是 P-Si2C4 与 P-C6 相比结构稳定的重要原因。更重要的是,Si 和 C 原子之间的屈曲随着锂的吸附而增加,在锂化状态 P-Li4Si2C4 下变为 0.876 Å。在这种状态下,SiC4 四面体的 C-Si-C(102.50° 和 124.59°)键角变得更接近标准四面体的 108.47°,表明 sp 3 -Si 原子的杂化和 C-Si 键的强度在 Li 吸附后变得更强。因此,随着锂吸附浓度的增加,12号带的分散也增强了,如图4所示。
当锂吸附在五硅石墨烯 (P-Si2C4) 表面时,电荷从锂原子转移(在实际电池运行中,Li + 离子来自内部电路,而相同数量的电子来自外部电路)到 P-Si2C4 中的碳原子。结果,多余的电子将沿着未占据的带(带 13 和 14 号)向下移动,从而产生系统的金属电子结构,如图 6b-d 所示。金属电子结构保证了P-Si2C4负极在充放电过程中良好的电子导电性,有利于P-Si2C4负极电池系统的倍率性能。
如上所述并由图 6 中的带投影电荷密度显示,带 No. 13 和 14 是 p P-Si2C4 中 C 原子的 z 态。这些空带对于锂吸附非常重要。除了带正电的锂离子和带负电的 P-Si2C4 衬底之间的库仑吸引力外,占据 C-p 的电子 z 态对带正电的 Si 原子具有很强的库仑吸引力(这会降低 13 和 14 号带的能级,从而降低衬底的总能量)。因此,P-Si2C4 上的锂吸附在能量上更有利。由于 P-Si2C4 的一个晶胞含有 4 个 C 原子,预计 4 个 Li 原子可以吸附在 P-Si2C4 表面。在 C-p 之后 z 态被完全占据,更多的锂原子在 P-Si2C4 上的吸附在能量上是不利的。这与图 4 中计算的吸附能一致。
P-Si2C4 上的锂离子迁移动力学
P-Si2C4 阳极的倍率性能由电子传导和锂离子扩散动力学决定。如上所述,虽然原始 P-Si2C4 的电子结构是绝缘体,但即使在锂浓度低时,它也会在锂吸附时自发地变成金属。因此,电子导电性应该足够好以用作负极材料。然后,锂离子在五硅石墨烯上的扩散成为速率控制步骤。由于 P-Si2C6 的结构类似于锂离子在其上扩散非常快的五石墨烯 (P-C6) [21],预计锂离子在 P-Si2C6 上的扩散也可以非常快.
电池的倍率性能与荷电状态 (SOC) 密切相关,即锂离子扩散取决于锂吸附浓度。为了评估不同SOC下的锂离子扩散动力学,考虑了两种极端浓度,即稀锂离子和稀锂空位。为了模拟稀锂离子扩散,一个锂离子被吸附在 P-Si2C4 的超级电池上。从上面“五硅石墨烯上的锂吸附”部分的讨论可知,当锂浓度较低时,锂离子更倾向于停留在 B1 位,如图 7 所示。考虑到结构的对称性,只有一个锂迁移路径(可以找到在图 7 中表示为 Path-1 的 Path-1,并且 Path-1 在 P-Si2C4 表面形成完整的 2D Li 扩散网络。 P-Si2C4 上沿 NEB 方法优化路径的稀锂离子迁移能垒约为 0.117 eV,小于 P-C6(0.17 eV,Path-II)[21] 和 g-Si2C4(0.548 eV) [13]。当SOC变为50%时,即材料放电进入Li2状态 Si2C4,所有锂离子仍占据 B1 位点(见图 3b),因此锂离子扩散路径与稀释锂离子的情况相同。这些结果表明,锂离子在放电过程的前半段扩散非常快。
<图片>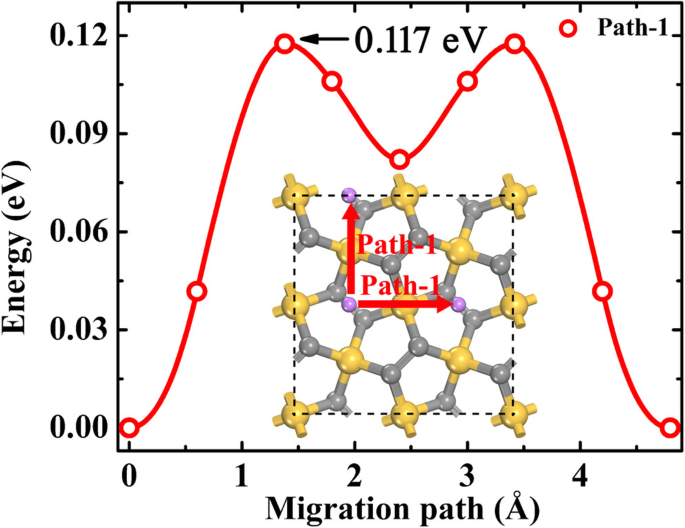
稀锂离子迁移路径和沿 P-Si2C4 路径的相应能量分布。灰色(中等大小)、金色和紫色球体分别是 C、Si 和 Li 原子。红色箭头表示二维扩散网络
对于稀锂空位的情况,我们从完全锂化状态的 Li4Si2C4 中去除一个锂离子,并在超级电池中创建一个稀锂空位。如上所述,当 Li 浓度高时,Li 离子更喜欢占据 H 位(见图 3c)。因此,考虑了三种不同的锂空位迁移途径,如图 8a 所示。路径-1 是指 Li 空位从 H 位点穿过 Si 原子顶部(跨 T 位点)迁移到相邻的 H 位点。 Path-2 对应于穿过顶层 C2-C2 二聚体中间点顶部的通路(穿过 B2 位点)。 Path-3 is the pathway along the C2–C2 dimer at the down-layer (across B1 site). The energy profiles along the optimized pathways are given in Fig. 8b. As is seen, the Li-ion migration energy barriers along these pathways are very low, particularly for Path-3 (0.052 eV). The energy profiles along Path-1 and Path-2 are slightly asymmetric, because of the large relaxation of the Li ions when one Li vacancy is created. The extremely low energy barrier along Path-3 is reasonable, since Path-3 crosses B1 site (energetically most favorable adsorption site). However, Path-3 alone is not able to form a complete Li diffusion network on the surface of the P-Si2C4. Therefore, Path-1 or Path-2 must take part in the diffusion process, and the overall energy barrier for dilute Li vacancy migration is 0.155 eV or 0.165 eV. Although higher than dilute Li-ion migration (0.117 eV), the energy barrier for dilute Li vacancy migration is also very small compared with that for P-C6 (0.25 eV, Path-II’) [21] and g-Si2C4 (0.233 eV) [13]. As the Li migration energy barriers in P-Si2C4 are always lower than those in P-C6 and g-Si2C4 (both dilute Li ion and dilute Li vacancy), it is expected that the rate performance of the P-Si2C4 is the best one among the three similar anode candidates.

一 Dilute Li vacancy migration pathways and b the corresponding energy profiles on fully lithiated P-Li4Si2C4. The gray (middle sized), golden, and purple spheres are C, Si, and Li atoms, respectively. The large green sphere represents the dilute Li vacancy. The thick/thin arrows indicate fast/slow migration pathways which form the two-dimensional diffusion networks
Conclusion
In summary, based on first principles calculations, we predicted that 2D pentagonal Si/C compound P-Si2C4 can be potentially used as anode materials for LIBs. Phonon dispersion data confirmed the dynamic stability of the P-Si2C4 structure at ground state, while AIMD simulation shows that the structure of the P-Si2C4 can be stable at temperatures as high as 2000 K. The unique 2D buckled pentagonal structure promotes special empty C-2p z states that facilitate Li adsorption on the surface of the P-Si2C4, which offers a gravimetric Li storage capacity of 1028.7 mAhg −1 . The calculated dilute Li-ion/Li vacancy migration energy barriers show that Li-ion diffusion on the surface of the P-Si2C4 can be faster than both the pentagonal graphene (P-C6) and the honeycomb-structured siligraphene. The metallic electronic structure of the lithiated P-Lix Si2C4 ensures good electronic conductivity of the material as electrodes. These advantages are crucial features to the P-Si2C4 as a promising anode material for LIBs. In summary, our first principles study offers a novel strategy to design high-performance Si/C complexity for the application in LIBs.
数据和材料的可用性
本研究期间生成或分析的所有数据均包含在这篇已发表的文章中。
缩写
- 二维:
-
二维
- AIMD:
-
Ab initio molecule dynamics
- bcc :
-
Body-centered face
- CINEB:
-
Climbing image nudged elastic band
- DFT:
-
密度泛函理论
- DOS:
-
态密度
- EVs:
-
Electric vehicles
- GGA:
-
General gradient approximation
- HSE06:
-
Heyd-Scuseria-Erznerhof
- LIB:
-
Li-ion batteries
- 爪子:
-
投影仪增强波
- PBE:
-
Perdew-Burke-Ernzerhof
- SOC:
-
State of charge
- VASP:
-
Vienna Ab initio Simulation Package
- vdW:
-
范德华
纳米材料
- Scalmalloy:用于金属 3D 打印的最新高性能材料
- 用于未来电池的锡纳米晶体
- 轻松合成锚定在 MWNT 上的 SiO2@C 纳米粒子作为锂离子电池的高性能阳极材料
- 少层二硫化钼/乙炔黑复合材料作为锂离子电池的高效阳极材料
- PPy 包覆的 MnO2 混合微材料的制备及其作为锂离子电池阳极的改进循环性能
- 不同粘合剂对锂离子电池金属氧化物阳极电化学性能的影响
- Na4Mn9O18/碳纳米管复合材料作为水性钠离子电池的高电化学性能材料
- 通过镁-热还原制备的嵌入式硅/石墨烯复合材料作为锂离子电池的阳极材料
- 3D 互连 V6O13 纳米片通过种子辅助水热工艺在碳化纺织品上生长,作为锂离子电池的高性能柔性阴极
- 通过脉冲激光沉积制备用于锂离子电池的纳米晶 Fe2O3 薄膜阳极
- CuGeO3 纳米线作为高级钠离子电池负极材料的合成和研究
- 氧化硅原位磁热还原制备的介孔硅微球用于钠离子电池中的高性能负极材料