

透明质酸功能化氧化钆纳米颗粒用于肿瘤的磁共振成像引导放射治疗
摘要
实体瘤的定位不准确和固有的放射抗性严重阻碍了放射治疗的临床实施。在这项研究中,我们通过一锅水热法制备了透明质酸功能化氧化钆纳米粒子 (HA-Gd2O3 NPs),用于有效的磁共振 (MR) 成像和肿瘤的放射增敏。由于HA功能化,所制备的直径为105 nm的HA-Gd2O3纳米颗粒在水中表现出良好的分散性、低细胞毒性和优异的生物相容性,并且很容易通过HA受体介导的内吞作用进入癌细胞的细胞质。重要的是,HA-Gd2O3 NPs 表现出高纵向弛豫率(r 1) 6.0 mM −1 S −1 作为 MRI 造影剂和放射增敏以剂量依赖性方式增强。这些发现表明合成的HA-Gd2O3 NPs作为双功能治疗诊断剂在肿瘤诊断和放射治疗中具有巨大的潜力。
介绍
放射治疗已广泛应用于癌症,其中涉及高能 X 射线和通过引起自由基损伤或 DNA 损伤在肿瘤部位沉积辐射剂量 [1,2,3,4]。然而,放疗敏感性差、肿瘤定位不准确、病灶区分能力差,以及正常组织引起的放疗副作用限制了放疗的临床实施[5]。因此,重要的是开发增强肿瘤放射敏感性同时最小化全身副作用的方法。纳米技术和放射治疗的结合是放射增敏的公认优先事项。
近年来,纳米技术被认为是一种有吸引力的癌症诊断和治疗策略 [6,7,8,9,10,11]。纳米粒子 (NPs) 的主要功能之一是基于纳米粒子通过被动靶向、增强渗透性和保留 (EPR) 效应、主动靶向和延长循环时间在肿瘤组织中选择性积累的精确肿瘤靶向 [12, 13,14]。我们之前的工作表明,用杂原子掺杂碳纳米粒子有效地调整了它们的内在特性并引入了有益的特征 [15,16,17]。有趣的是,NPs 可以用作放射增敏剂 [18]。重金属(具有高 Z 元素)NP(例如,Au、Bi、Gd)作为有前景的放射增敏剂可用于放射增敏治疗,因为它们具有高 X 射线光子捕获截面和康普顿散射效应 [19]。当 X 射线与高 Z 纳米粒子、俄歇电子和光电子相互作用时,二次电子的释放会伤害癌细胞,从而在放射治疗期间增加剂量 [20]。迄今为止,钆基纳米颗粒 (GdNP) 已被证明是有效的 MRI 造影剂 (CA) [21, 22],并以无创方式实时区分正常组织与病变组织和病变。这些试剂缩短纵向弛豫时间以影响纵向弛豫r 1 [23],并且他们不准确的定位经常导致副作用。众所周知,透明质酸(HA)是一种主要的配体和药物递送载体,可靶向具有簇决定簇44(CD44)等HA受体的位点[24,25,26]。
基于先前的研究结果,我们使用 HA 作为靶向配体来功能化 Gd2O3 NPs,具有双重功能:有效的肿瘤靶向 MRI CA 和放射增敏剂,以克服固有的放射抗性和肿瘤定位的不准确性。此外,HA-Gd2O3 NPs 显示出高纵向弛豫率 (r 1 ) 作为具有更好 MR 成像质量的有前途的 MR 显像剂。与目前可用的 CAs [27, 28] 相比,由此产生的 HA-Gd2O3 NPs 显示出三个显着优势:首先,由于使用天然细胞外基质作为前体,HA-Gd2O3 NPs 表现出良好的生物相容性。其次,HA功能化显着提高了肿瘤靶向性并减少了副作用。最后,HA-Gd2O3 NPs 在诊断和治疗方面具有双功能能力。为了验证增强放射增敏的有效性并探索其机制,我们评估了HA-Gd2O3 NPs对肿瘤细胞活力、细胞周期和细胞凋亡的放射增敏作用。
结果与讨论
HA-Gd2O3 NPs 的制备和表征
在这项研究中,HA-Gd2O3 NPs 使用简单的水热工艺成功制备,如方案 1 所示。通过动态光散射分析粒径,而通过透射电子显微镜进行形态检查。如图 1a 所示,HA-Gd2O3 NPs 表现出均匀的分散和离散的准球形形状,没有明显的聚集。 HA-Gd2O3 NPs 的平均直径为 105 nm。与正常组织相比,肿瘤组织的毛细血管内皮通透性增加,内皮间隙为100-600 nm[29]。因此,具有理想尺寸的纳米颗粒很容易浸入肿瘤组织中,它们可能会显着提高被动靶向给药的效率。
<图片>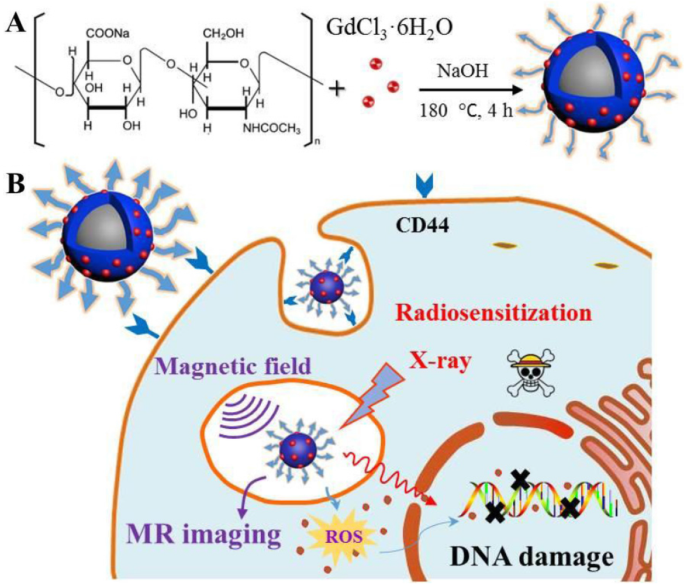
水热法合成HA-Gd2O3 NPs的示意图(A)和以下生物医学应用(B)

HA-Gd2O3 NPs 的表征。低 (a ) 和高 (b ) 放大 TEM 图像。直径分布 (c ) 和 XRD 图 (d ) HA-Gd2O3 NPs
HA-Gd2O3 NPs 的相结构通过 XRD 研究,使用氧化石墨 (GO) 功率作为对照。如图 1d 所示,一个主衍射峰 (2θ =12.04°) 和两个小峰 (2θ =29.6°, 42.1°) 在 HA-Gd2O3 NP 衍射图中观察到,分别对应于 GO(100 面)和石墨(002 面)的特征峰。 HA-Gd2O3 NPs的主晶格间距比d的GO间距显示出更小的间距(由布拉格公式计算为0.73 nm) 001 =0.85 nm,其中峰值位置的向上移动可能归因于 sp 3 之间的间距减小 层。所有宽衍射峰表明HA-Gd2O3 NPs具有无定形结构,这可能归因于高度无序的碳和sp 2 (C-C) 碳化过程中的层间距。结果进一步表明结晶性差的HA-Gd2O3纳米颗粒具有异质多层结构,这与我们之前关于碳点的报道一致[30, 31]。
HA-Gd2O3 NPs 的化学结构和表面组成
使用傅里叶变换红外光谱和 X 射线光电子能谱研究了 HA-Gd2O3 NPs 的表面官能团和组成。如图 2a 所示,XPS 光谱在 284.0、400.0、530.6 和 1188.5 eV 处出现四个典型峰,表明 HA-Gd2O3 NPs 主要由钆、碳、氧和氢元素组成。 Gd3d的高分辨率光谱 (图2b)揭示了在1187.5 eV和1221 eV处存在两个强峰,对应于32 eV的自旋轨道分裂,分别对应于3d 5/2 和 3d Gd 的 3/2 能级。这些观察结果与之前关于 HA-Gd2O3 NPs 的报道非常一致。 O (1s ) 图 2c 中显示的光谱由位于 531.4 eV 的一个主峰支配,它对应于 O 2− 之间的键 和 Gd 3+ .对裸露和 HA-Gd2O3 NP 样品进行了 FITR 光谱分析(图 2d)。对于裸样品,如图 2a 所示,v 的特征吸收带 作为 O-C-O 在 1580 和 1380 cm −1 揭示了碳酸酯基团的存在。在 3340 和 2900 cm −1 处的宽峰 分别归因于 O-H 和 C-H 伸缩振动,它们对应于表面吸附的水。对于 HA-Gd2O3 NPs,COO − 的伸缩振动 在 1580 和 1380 cm −1 增强,表明引入了透明质酸的羧基。这些结果表明,HA-Gd2O3 NPs 的官能团主要含有一定数量的羰基、羧酸根和羟基。位于表面的这些官能团的存在赋予了 HA-Gd2O3 NPs 在水中优异的分散性。重要的是,嵌入纳米颗粒表面的Gd离子可用于MR成像和放射增敏,这极大地防止了Gd离子泄漏到周围环境中。
<图片>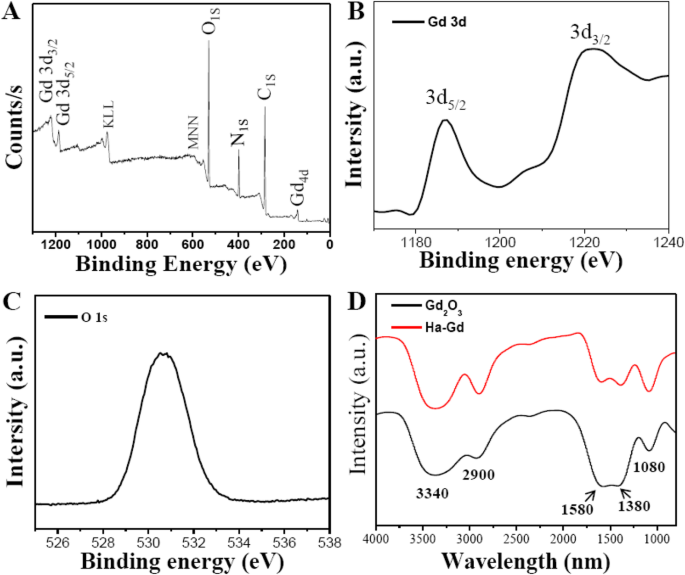
HA-Gd2O3 NPs 的化学结构和组成。 一 HA-Gd2O3 NPs 的全扫描 XPS 光谱。 b Gd3d 光谱。 c O1S 光谱。 d HA-Gd2O3 NPs的FTIR光谱
HA-Gd2O3 NPs 的生物相容性
HA-Gd2O3 NPs 作为潜在的生物医学制剂,其生物相容性对其生物医学应用至关重要。首先,使用 CCK-8 测定法在 HepG2 和 VSMC 细胞中评估了 HA-Gd2O3 NPs 的固有细胞毒性。如图 3a 和 S 所示。图 1,HA-Gd2O3 NPs 在长达 3 天的时间内没有表现出明显的细胞毒性。即使在 24 h 暴露时间后浓度为 200 μg/mL,细胞活力仍约为 90%。其次,HA-Gd2O3 NPs 在体内的血液相容性使用溶血试验进行评估。如图 3b 所示,我们观察到水中红细胞的溶血(阳性对照)。相比之下,HA-Gd2O3 NP在0至200 μg/mL的不同浓度下孵育2 h后未观察到明显的溶血,这与PBS溶液(阴性对照)的结果相似。与阴性对照相比,根据上清液在 541 nm 处的吸光度,略微评估了不同浓度 HA-Gd2O3 NPs 的溶血百分比。结果表明,HA-Gd2O3 NPs 在研究浓度下的溶血率均小于 3%,验证了其良好的血液相容性。为了检查潜在的毒性,将 HA-Gd2O3 NPs 静脉注射到 Balb/c 小鼠中(如图 2 所示)。 1周后处死小鼠,取膀胱、肾、脾进行病理切片分析。如图2所示,病理切片结果显示HA-Gd 2 O 3 NP处理的小鼠器官中没有可观察到的病变或炎症反应。这些结果清楚地表明合成的HA-Gd2O3 NPs具有良好的细胞相容性和良好的血液相容性。
<图片>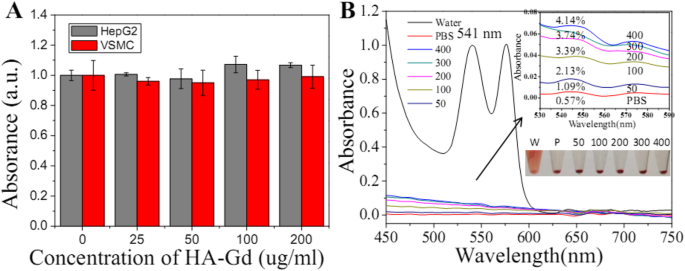
一 使用 CCK-8 测定,HA-Gd2O3 NPs 对 HepG2 和 VSMC 细胞活力的影响。 b HA-Gd2O3 NPs 在不同浓度(50、100、200、300 和 400 μg/mL)下的溶血活性。 PBS和水分别作为阴性和阳性对照
MRI 幻像研究
纵向 (T 1) HA-Gd2O3 NPs 的弛豫时间与商业造影剂 Magnevist (Gd-DTPA) 一起使用磷酸盐缓冲盐水 (pH =7.4, 0.2 M) 作为对照进行了体外研究。随着 Gd 浓度的增加,T 的信号强度 1权重幻影图像明显增加,表明所有样本都可以在T上产生MRI对比度增强 1 加权序列(图 4a)。此外,信号强度与反转时间的关系图给出了 T 1-特定浓度下每种造影剂的松弛时间。如图 4b 所示,r 1 HA-Gd2O3 NPs 的测量值为 6.0 mM -1 S −1 , 明显高于 Magnevist (3.86 mM −1 S −1 ) 相同条件下。增强的 r 1 可能归因于 HA-Gd2O3 NPs 更高的流体动力学半径和表面积;因此,更多的掺杂在纳米颗粒晶格中的 Gd 原子变得可接近水分子,缩短纵向弛豫并增强 r 1 价值。
<图片>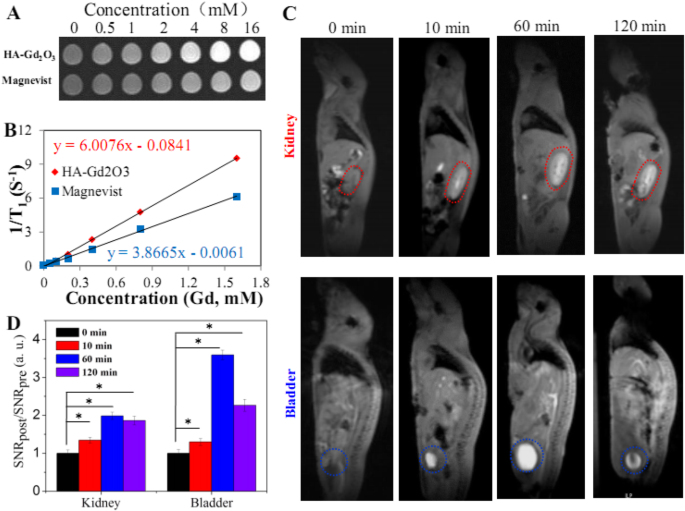
一 T1 具有不同总 Gd 离子浓度的 HA-Gd2O3 NPs 的幻影图像。 b 1/T1 的对应图 对总 Gd 离子的浓度。 c 体内 T1 静脉注射 HA-Gd2O3 NPs 作为造影剂后小鼠的 MR 成像和分析。 d 给药后不同时间点膀胱和肾脏信号变化(SNR比)的量化(n =3)。 *p <0.05
体内磁共振成像
为了确定体内的潜在应用,以正常 BALB/c 小鼠为模型,使用 MRI 扫描仪研究了 HA-Gd2O3 NPs(10 mg/kg)的 MR 成像性能和循环命运。与注射前图像(图 4c)相比,在注射后 10 min 时可以清楚地观察到如虚线圆圈所示的肾脏和膀胱的较亮区域,表明 HA-Gd2O3 NPs 可用作 CAs 增强 T 1 体内这些主要器官的松弛。重要的是,HA-Gd2O3 NPs 的对比度增强保持在注射后 60 min,远长于 Gd 复合小分子(在小动物中的半衰期约为几分钟)[27],表明 HA-与商业对比剂相比,Gd2O3 NPs 在体内具有更长的保留时间。为了定量分析 MR 对比效应,我们通过精细分析 MRI 图像的感兴趣区域来计算信噪比(SNR),并计算 SNRpost/SNRpre 的值来表示相对信号增强(RSE)(图 3)。 4d)。肾脏中 HA-Gd2O3 NPs 的 RSE 值为 1.35、1.99 和 1.86。同样,膀胱中 HA-Gd2O3 NPs 的 RSE 值高于肾脏(1.29、3.59 和 2.26)。大尺寸(超过 100 nm)的 HA-Gd2O3 NPs 显示出较长的循环时间,根据之前的结果,可以通过胆肠途径消除 [32]。此外,体内循环时间的延长可能通过增强EPR效应增加了肿瘤的被动靶向作用。
肿瘤成像
具有优异 MR 成像性能和长循环时间的 HA-Gd2O3 NPs 为通过 EPR 效应对肿瘤进行成像提供了很好的机会。因此,我们建立了皮下肝肿瘤模型,以研究是否可以通过 HA-Gd2O3 NP 增强 MRI 检测到肝细胞癌 (HCC)。在静脉注射 HA-Gd2O3 NPs 后,我们观察到皮下肿瘤区域变得比周围组织更亮,如冠状和横向扫描所见(图 5a)。皮下肿瘤的 RES 在横向和冠状扫描时分别显着达到 1.54 和 1.83,表明 HA-Gd2O3 NPs 通过 EPR 效应在肿瘤区域有效积累。这些发现表明 HA-Gd2O3 NPs 对肿瘤的 MRI 可视化高度敏感。许多研究表明,较长的循环时间可以提高实体瘤渗漏血管被动靶向的效率[33]。
<图片>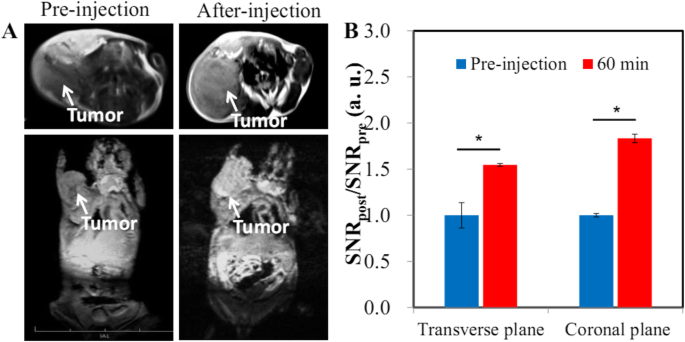
T 1 加权 MR 成像 (a ) 和相应的定量分析 (b ) HA-Gd2O3 NPs静脉注射后Heps小鼠肝癌异种移植瘤
HA-Gd2O3 NPs 的辐射增敏作用
HA-Gd2O3 NPs 作为 T 的优异性能 1 造影剂促使我们准确地确定其在肿瘤放射增敏作用中的位置。首先,使用 CCK-8 试验探索了这种联合治疗是否发生了剂量增加。如图 6a 所示,单个 HA-Gd2O3 NPs(浓度 0-200 μg/mL)没有显着影响 HepG-2 的活力,但 X 射线(范围 0-9 Gy)照射降低了 HepG-2 细胞的活力9 Gy 时低于 70%。重要的是,X 射线照射与 HA-Gd2O3 NPs 的组合使 HepG-2 细胞活力显着降低至不到 50%,特别是在 200 μg/mL 的 9 Gy 浓度下。接下来,进行克隆测定以评估 X 射线照射和 HA-Gd2O3 NPs 组合后 HepG-2 细胞中的细胞活力。如图 6b 所示,单个 HA-Gd2O3 NPs 对 HepG-2 细胞集落形成没有显着影响,但 X 射线照射使 HepG-2 细胞集落形成减少到 46.7%。然而,用 X 射线照射和 HA-Gd2O3 NPs 处理显着抑制了 29.8% 的细胞集落形成活力。相应的图像进一步验证了HA-Gd2O3 NPs对HepG-2细胞的放射增敏作用。
<图片>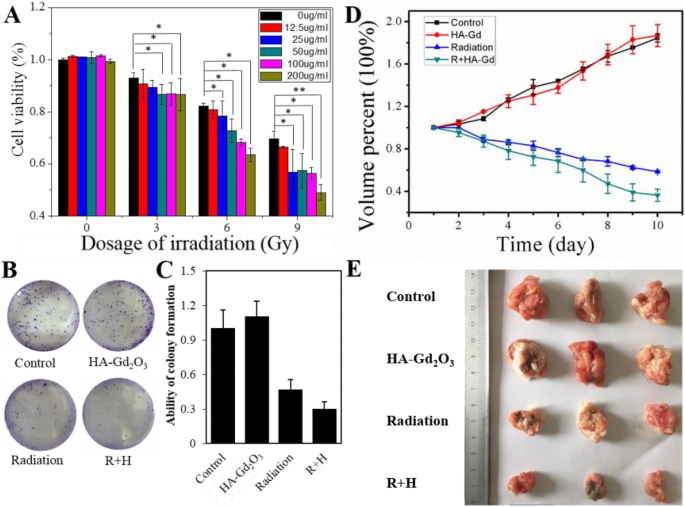
HA-Gd2O3 NPs 的体外放射增敏作用。 一 HA-Gd2O3 NPs(0 μg/mL、12.5 μg/mL、25 μg/mL、50 μg/mL、100 μg/mL和200 μg/mL)与X射线辐射(0 Gy)的协同作用Gy、6 Gy 和 9 Gy)对使用 CCK-8 测定的 HepG2 细胞活力的影响。 *p <0.05, **p <0.01,差异有统计学意义。 b HA-Gd2O3 NPs和X射线辐射和c处理HepG2细胞的集落存活分析 相应的克隆生成能力定量分析。 d 不同组小鼠不同治疗后肿瘤体积生长曲线。组:a 控制,b 辐射,c HA-Gd2O3 NPs 和 d 辐射 + HA-Gd2O3 NPs。 e 不同组小鼠治疗结束时肿瘤照片
体外放疗的有效性鼓励我们应用 HA-Gd2O3 NPs 结合照射来控制荷瘤小鼠的肿瘤生长。对照和单一 HA-Gd2O3 NP 治疗中的肿瘤体积增长迅速,均增加了 180%。与 58% 的单次照射治疗相比,HA-Gd2O3 NPs 联合放射治疗在照射 10 天后显示出 38% 的有效肿瘤抑制(图 6d)。对肿瘤进行拍照,各组平均肿瘤体积显示,在所有组中,(a)组的平均肿瘤体积最大,而(d)组的平均肿瘤体积最小(图6e)。这些结果表明所制备的HA-Gd2O3 NPs有望在X射线照射下抑制肿瘤生长。
为了进一步了解HA-Gd2O3 NPs和X射线辐射联合治疗的放射增敏机制,进行了流式细胞术测定和活/死染色测定。如图 7a 所示,在用单一 HA-Gd2O3 NPs 处理的 HepG-2 细胞中观察到强烈的绿色荧光而没有红色,这证实了高细胞活力。 X射线辐射处理后观察到少量红色荧光,表明细胞凋亡水平较低。然而,在 HA-Gd2O3 NPs (200 μg/mL) 和辐射联合处理后观察到强烈的红色荧光,表明 HA-Gd2O3 NPs 可以显着增强 X 射线辐射诱导的细胞凋亡。使用流式细胞术进一步研究了 HA-Gd2O3 NPs 的放射增敏机制。如图 7b 所示,单次辐射和 HA-Gd2O3 NPs 分别诱导了 27.55% 和 19.12% 的 G2/M 相停滞。然而,HA-Gd2O3 NPs 和辐射的联合治疗显着增加了 G2/M 期停滞,以剂量依赖性方式从 30.89% 到 33.27%。同时,联合治疗诱导了 10.63% 至 22.67% 的细胞凋亡,这反映在单个 HA-Gd2O3 NPs 和辐射诱导 3.02% 和 13.87% 时的亚 G1 比例。为了进一步研究细胞死亡的可能机制,通过流式细胞术进行了膜联蛋白 V-EGFP/PI 方法。膜联蛋白 V-EGFP 发射信号绘制在 x -轴,而 PI 发射信号绘制在 y 上 -轴(图 8)。坏死细胞、早期凋亡细胞、晚期凋亡细胞和活细胞的数量由Annexin V - 的百分比决定 /PI + (Q3), Annexin V + /PI + (Q2), 和 Annexin V − /PI − (Q4),分别。对照组和单一HA-Gd2O3 NP(100 μg/mL)组细胞凋亡率均小于5%,影响不大。与单一辐射治疗组(8.8%)相比,HA-Gd2O3 NPs与辐射联合治疗的细胞凋亡率随着浓度的增加而增加,范围为50~200 μg/mL。特别是当浓度为200 μg/mL时,细胞早期凋亡率明显提高至33.2%,早期和晚期凋亡率达到44.3%。一般来说,HA-Gd2O3 NPs和X射线照射的组合对细胞集落形成、细胞活力呈剂量依赖性和放射增敏作用具有协同作用。基于这些结果,HA-Gd2O3 NPs可能是增强放射治疗放射增敏的有效替代方法。
<图片>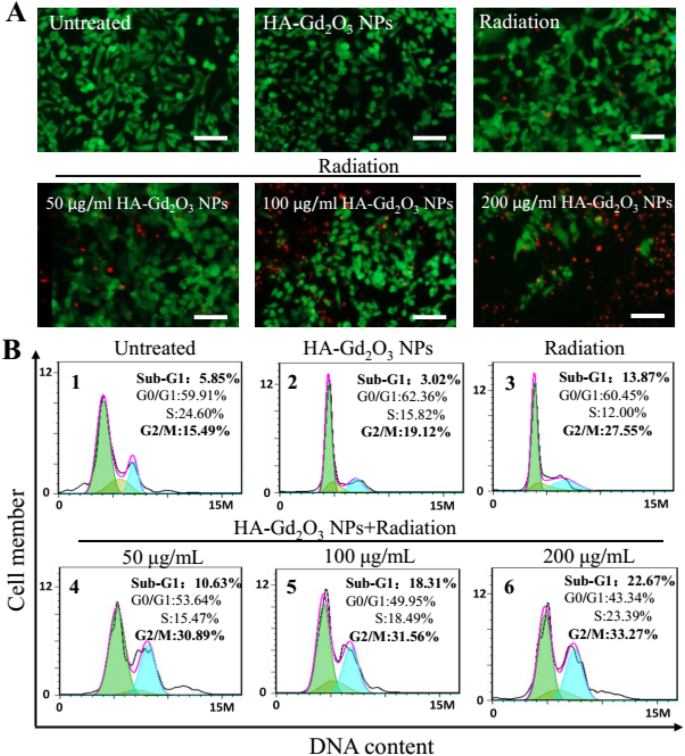
HA-Gd2O3 NPs 的辐射剂量增强。 一 HepG2 细胞的活死染色。绿色(双乙酸荧光素染色)=活细胞,红色(碘化丙啶染色)=死细胞。比例尺 =200 mm。 b 流式细胞术定量DNA含量分析不同处理后细胞周期分布
<图片>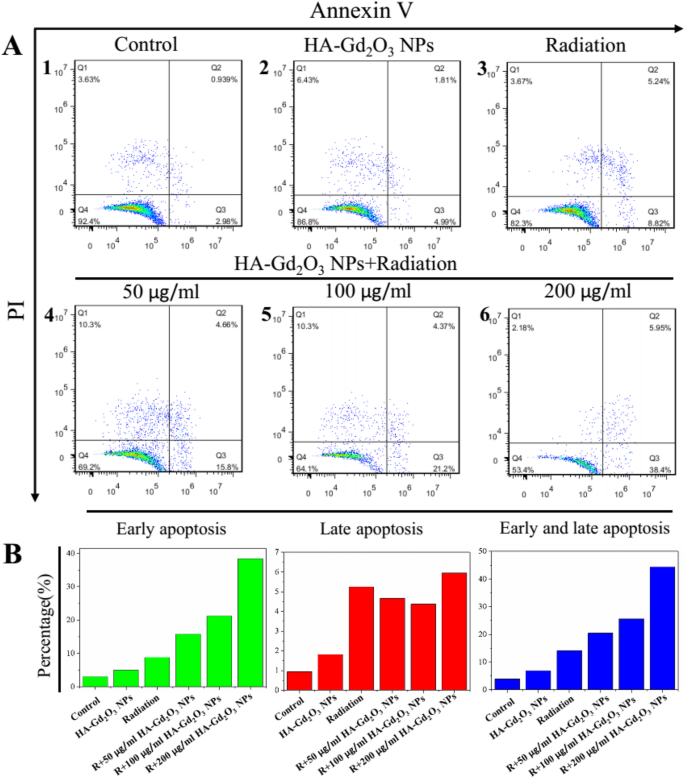
HA-Gd2O3 NPs 的凋亡分析。 一 X射线照射后24 h时HepG-2细胞的凋亡水平。 b 相应的定量分析
结论
总之,我们开发了一种双功能 HA-Gd2O3 NPs,用于在一锅热液过程中对肿瘤进行有效的 MR 成像和放射增敏。用HA包覆后,合成的HA-Gd2O3 NPs在水中表现出良好的分散性和良好的生物相容性。所得到的包裹 Gd 原子的 HA-Gd2O3 NPs 不仅有效地表现出高纵向弛豫率 (r 1 ) 对于 MRI 作为 T 1 造影剂,但也通过作为放射增敏剂诱导细胞凋亡和阻断细胞周期来增强肿瘤的放射敏感性。因此,新型HA-Gd2O3 NPs在肿瘤诊断和放射治疗方面具有广阔的应用前景。
材料和方法
材料
荧光素二乙酸酯 (FDA) 和碘化丙啶 (PI) 购自 Sigma(纽约,纽约,美国)。 GdCl3·6H2O、乙二醇(99%)购自恒瑞药业有限公司(江苏连云港)。 Cell Counting Kit-8 (CCK-8) 购自 Dojindo (Kumamoto, Japan)。 NaH2PO4、Na2HPO4 和 H2SO4 购自光复精细化工研究所(中华人民共和国天津南开)。胎牛血清和 Dulbecco 最低必需培养基(DMEM)购自 Invitrogen 中国有限公司(中华人民共和国上海)。所有化学品均为分析纯,无需进一步纯化即可使用。
HA-Gd2O3 NPs 的合成
HA-Gd2O3 NPs 使用一锅水热法制备如下:首先,将 0.1 g HA 溶解在 20 mL 水中,在环境温度下剧烈搅拌过夜。随后,分别加入 0.1 g GdCl3·6H2O 和 0.5 mL NaOH (1 M)。然后,将混合物再搅拌 5 分钟以形成均匀的澄清溶液并转移到 50-mL 高压釜中,密封,并在 120 °C 下水热处理 6 小时。自然冷却至室温后,透明悬浮液用0.22-μm膜过滤以去除任何大尺寸的团聚体。然后将制备的溶液在具有 14-kDa 截留分子量的透析袋中用水透析 3 天。收集透析液并使用真空冷冻干燥机冷冻干燥。由此获得HA-Gd2O3 NP粉末并保存用于进一步表征。
检测和表征
在 200 kV 的加速电压下,在 JEM-2100 显微镜(JEOL,Tokyo,Japan)上通过高分辨率透射电子显微镜检查 HA-Gd2O3 NPs 的形态。 HA-Gd2O3 NPs 的元素组成通过 MK II 光电子能谱仪中的 XPS 测量确定,使用 Al-Ka (1486.6 eV) 作为 X 射线源和傅里叶变换红外 (FTIR) 光谱仪 (Nicolet Nexus 470, GMI, Ramsey ,明尼苏达州,美国)。 HA-Gd2O3 NPs 的晶体结构在配备 Cu Kα (λ =0.15405 nm) 辐射,扫描速度为 4°/min,范围为 5 到 80°。
细胞活力测定
通过细胞计数试剂盒 CCK-8 测定(CCK-8 测定)研究 HA-Gd2O3 NPs 对细胞活力的影响。 HepG2(人肝癌,ATCC 编号:HB-8065)和 VSMC(血管平滑肌细胞)以 3 × 10 3 的密度接种在 96 孔板中 细胞/孔,在 37 °C 的 5% CO2 培养箱中培养 24 h,然后使用含有不同浓度 HA-Gd2O3 NPs(0 μg/mL、25 μg/mL、50 μg/mL、100 μg/mL、和 200 μg/mL) 代替生长培养基。再孵育4 h后,每孔加入10μL CCK-8溶液,避光孵育4 h。使用 Synergy HT Multi-Mode Microplate Reader (Bio Tek, Winooski, VT, USA) 在 490 nm 处测量吸光度。未处理的细胞(在 DMEM 中)用作对照,相对细胞活力(平均值 SD,n =5) 表示为 Abssample/Abscontrol × 100%。
溶血分析
简而言之,19-21g,6 周龄雌性 BALB/c 小鼠由中华人民共和国卫生部动物管理条例友情制备。从眼球中取出三毫升用肝素钠稳定的血液。然后按文献1200 rpm、15 min离心除去上清液。之后用PBS洗涤沉淀5次,得到小鼠红细胞(MRBCs),然后取出眼球取约3mL血液,用肝素钠稳定,离心(1, 200 rpm, 15 min)按文献[27]除去上清液,用PBS洗涤5次,得到小鼠红细胞(MRBCs)。用 PBS 稀释 10 倍,将 0.1 mL MRBCs 转移到 1.5 mL 管中,该管预装了 0.9 mL PBS,其中含有不同颗粒浓度(50-200 μg/mL)的 HA-Gd2O3 NPs、0.9 mL 水(作为阳性对照)和 mL 0.9 mL PBS。 PBS(作为阴性对照),分别。轻轻摇动后,将混合物在室温下孵育 2 小时,然后以 12,000 rpm 离心 1 分钟。最后,拍摄所有样品的照片,并通过 UV-2450 紫外-可见分光光度计测量上清液(血红蛋白)的吸光度。将样品、阳性、阴性对照在541 nm处吸光度的差值除以不同样品的溶血百分比。
体外和体内 MR Phantom 研究
在 3 Tesla MRI 扫描仪(Magnetom Trio Tim,Siemens,Germany)上获取体外和体内 MR 图像。研究T 在体外体模图 1 中,将 0 到 16 mM 不同浓度的 HA-Gd2O3 NPs 溶液添加到 96 孔培养板中,使用 Magnevist(商用 MR 造影剂,Gd-DTPA)作为对照。对于体内 MR 成像,我们选择正常 BALB/c 小鼠作为模型 (n =4)。动物实验严格按照中华人民共和国卫生部动物管理规定进行。通过无菌膜过滤器(孔径 0.22 μm)过滤的每公斤 10 毫克 HA-Gd2O3 NPs 被静脉注射到动物体内,然后立即用 MRI 扫描仪进行研究。这些用于体外 MR 成像和体内动物的样品使用以下参数进行成像:TR/TE =300/10 ms,256 × 256 矩阵,切片 =5,厚度 =2 mm,平均值 =2,FOV =80 × 80 .
体外辐射增敏作用
CCK-8 assay was used to evaluate radiosensitizing activity of HA-Gd2O3 NPs in vitro. Cells were seeded in five 96-well plates at a density of 3 × 10 3 cells/well, and each plate was treated at the same condition:cultured at 37 °C in 5% CO2 incubator for 24 h, then using DMEM containing different concentrations of HA-Gd2O3 NPs (0 μg/mL, 12.5 μg/mL, 25 μg/mL, 50 μg/mL, 100 μg/mL, and 200 μg/mL) replacing the growth medium. After incubation for 4 h, five plates were irradiated at different X-ray doses (0 Gy, 3 Gy, 6 Gy, and 9 Gy), 300 cGy/min, respectively. After radiation, all the plates were incubated for 4 h, then measuring the absorbance under the same parameters.
Clonogenic survival assay was also conducted to study the radiosensitization effect in vitro. HepG2 cells were seeded in six-well plates at 4.0 × 10 4 cells/well and allowed to grow for 16 h. The cells were incubated with HA-Gd2O3 NPs diluted in cell culture medium for 6 h. The cells were then irradiated at 6 Gy using a clinical linear accelerator (Oncor, Siemens, Germany) with 6 MeV irradiation using a 10 cm × 10 cm radiation field at a source-to-skin distance (SSD) of 100 cm to cover the entire cells. Then, these irradiated cells were allowed to grow for 14 days, fixed with 4% paraformaldehyde at room temperature for 40 min, stained with a 1% crystal violet after washing the cells.
Live-Dead Staining Assay and Flow Cytometry
To study the radiosensitization effect of HA-Gd2O3 NPs, HepG2 cells were seeded in six-well plates at a density of 4.0 × 10 4 cells/well and allowed to grow for 12 h and set six groups (control, HA-Gd2O3 NPs, radiation, radiation + 50 μg/mL HA-Gd2O3 NPs, radiation + 100 μg/mL HA-Gd2O3 NPs and radiation + 200 μg/mL HA-Gd2O3 NPs). When cells were grown to 80% in plates, the first group had no treatment, the second group was incubated with 100 μg/mL HA-Gd2O3 NPs for 24 h, the third group was just irradiated, and the fourth group to the sixth group were irradiated and incubated with different concentrations of HA-Gd2O3 NPs (50, 100, and 200 μg/mL) for 24 h, respectively. After that, FDA and PI working buffer were added for cell staining. The fluorescence of stained cells was observed under a fluorescence microscope (×20), and live cells showed green color and dead ones exhibited red color. Furthermore, cells by different treatments were washed three times with PBS, digested, collected, and centrifuged at a speed of 2000 rpm for 5 min, then fixed with 70% ethanol at − 20 °C overnight followed by PI staining. DNA fragmentation was quantified by the fluorescence intensity of PI on a flow cytometer (BD, Accuri, C6BD, Accuri, C6), analyzed by software (FLOWJO 7. 6. 2) to make clear the cell cycle distribution.
Radiosensitization Effect In Vivo
Female BALB/c mice with body weights of 19–21 g and ages of 6 weeks were obtained from the Yangzhou University Laboratory Animal Center under the standard conditions. Animal experiments were compliant with the Animal Management Rules of the Ministry of Health of the People’s Republic of China. A subcutaneous tumor model was established as the following procedures:First, 1 × 10 6 HepS cells were inoculated into mice intraperitoneal, and the ascites were collected after 5 days. Then, these ascites were injected into subcutaneous. When the tumor sizes reached approximately 100 mm 3 , subcutaneous tumors models were established and applied to the following experiments.
Eight mice bearing subcutaneous tumors per group were treated with radiation at 3 Gy per fraction to a total dose of 9 Gy within 7 days. The radiotherapy was conducted after 3 h intravenous injection of HA-Gd2O3 NPs (10 mg/Kg), on a Siemens Primus clinical linear accelerator (6 MeV) using a self-made device to cover the entire tumor. These mice were anesthetized by 1% pentobarbital (50 mg/kg) to assure immobility during irradiation. The volume was measured and recorded every day, determined from caliper measurement, and calculated by the formulae:V =0.5 × a × b 2 , 其中 V (mm 3 ) is the volume of the tumor, and a (mm) and b (mm) are the tumor length and tumor width, respectively. Relative tumor volumes were normalized to their initial sizes. Each group was conducted on eight mice, wherein statistical analysis was performed using Student’s two-tailed t test (*p <0.05, **p <0.001).
数据和材料的可用性
The data sets supporting the results of this article are included within the article.
纳米材料
- 用于化学传感器的金纳米粒子
- 用于磁性传感器的纳米金刚石
- 用于癌症治疗的纳米粒子:当前的进展和挑战
- 钴掺杂 FeMn2O4 尖晶石纳米粒子的制备和磁性
- 铁电纳米粒子中的渗透磁性
- 磁性纳米粒子组装中的相互作用
- 使用聚(4-苯乙烯磺酸-共-马来酸)增强金磁性纳米颗粒的稳定性:用于蛋白质检测的定制光学特性
- 改性超支化聚甘油作为分散剂,用于控制和稳定碳氢化合物中的金纳米粒子
- 用于光热疗法和光声成像的聚吡咯涂层铁铂纳米粒子的合成和体外性能
- 基于叶酸和 gH625 肽的 Fe3O4 磁性纳米颗粒功能化增强细胞内化的比较
- 磁性金纳米粒子标记乙酰肝素酶单克隆抗体及其在肿瘤磁共振成像中的后续应用
- 通过腹膜内和静脉内给药途径对大鼠生物合成铜和氧化锌纳米颗粒的体内比较检查