

用于神经保护的脑靶向聚山梨醇酯 80 乳化多奈哌齐载药纳米颗粒
摘要
由于血脑屏障,大多数阿尔茨海默病药物无法有效发挥作用。因此,我们设计了一种新的纳米制剂(PS-DZP-CHP):具有聚山梨醇酯80(PS)表面覆盖的胆固醇修饰的支链淀粉(CHP)纳米颗粒,作为多奈哌齐(DZP)载体实现脑组织递送。通过尺寸分析和等温滴定量热法,我们选择了药物与纳米材料(1:5)的最佳给药比例,并设计了一系列实验来验证纳米颗粒的功效。体外释放实验结果表明,纳米颗粒可以在 72 小时内实现持续药物释放。小鼠荧光观察结果表明 PS-DZP-CHP 纳米粒子具有良好的脑靶向性。此外,纳米颗粒可以提高小鼠脑组织中的药物浓度。 DZP-CHP纳米颗粒用于预处理具有Aβ蛋白损伤的神经细胞。 MTT、罗丹明123和AO-EB染色法测定乳酸脱氢酶浓度,证明DZP-CHP纳米粒对Aβ25-35诱导的神经毒性具有保护作用,优于游离多奈哌齐。微热永动机测试表明,PS-DZP-CHP纳米粒子与载脂蛋白E具有亲和力,这可能是该纳米粒子靶向脑组织的关键。
介绍
AD是一种病理机制复杂的中枢神经系统疾病,会导致进行性认知功能障碍,但由于给药难度大、可到达脑组织的药物浓度低,治疗难度很大[1, 2]。 BBB 的通过已成为给药系统 (DDS) 研究中的一个主要困难 [3, 4]。大多数药物通过生物、化学或物理方式打开 BBB;其中,目前临床上最常用的是物理方法[5]。由于基于侵入性技术的颅内给药存在无法克服的缺陷,通过对药物表面的化学修饰来制备亲脂性前药或主动转运底物更具优势[6]。其中,纳米颗粒是通过主动靶向血脑屏障实现颅内给药的最佳选择之一[7,8,9]。
纳米 DDS 已成为大脑靶向研究的焦点,包括聚合物纳米粒子、有机纳米粒子、脂质体、纳米纤维和胶束,旨在提供治疗和诊断 [6, 10,11,12]。载药纳米颗粒和小分子亲脂性药物均可通过血脑屏障。不同之处在于,载药纳米颗粒更有可能通过吸附在大脑毛细血管壁上而通过被动扩散。此外,游离药物面临第二道屏障,即血脑脊液屏障 (B-CSF) [13]。与其他屏障对应物不同,大多数药物相对可渗透脑脊液并扩散到脑实质中。但由于扩散过程非常缓慢,CSF中游离药物的浓度远高于脑实质,CSF中的高浓度会引起一定的毒性[14, 15]。在这个过程中,纳米粒子具有独特而显着的优势。如果纳米载体表面涂有亲水性表面活性剂,ApoE会吸附到表面,CSF可以促进纳米颗粒通过血管周围的空间向脑实质移动[16]。由 Kreuter 等人制备的非离子表面活性剂聚山梨醇酯 80 包覆的纳米颗粒。 [17] 是第一个成功输送到大脑系统并通过血浆中血清蛋白 ApoE 吸附转运的药物。因此,在纳米颗粒表面修饰聚山梨醇酯80形成药物特异性复合物,可以靶向药物和内源性BBB受体识别,大大提高药物的生物利用度[18,19,20]。
目前,乙酰胆碱酯酶 (AChE) 抑制剂多奈哌齐 (DZP) 常用于治疗中度 AD,但由于其脂溶性,体内 溶出度差 和口服生物利用度低,必须每天服用常规多奈哌齐片以维持治疗效果 [21, 22]。由于AD患者的认知能力严重受损,给维持用药计划带来很大的不便,开发长效缓释DZP制剂迫在眉睫。淀粉样蛋白级联假说表明 AD 的原因是淀粉样蛋白斑块在脑实质和脑血管壁周围的沉积。在脑区也观察到大量弥漫性斑点,这些斑点由无定形、高度纤维化和不溶性细胞外 Aβ 沉积物组成 [23, 24]。鲍里迪等人。发现无毒、易降解的支链淀粉多糖纳米粒可与Aβ蛋白形成复合物,有效阻止蛋白聚集,并可快速从细胞中清除,抑制细胞毒性[25]。胆固醇疏水改性支链淀粉 (CHP) 是一种两亲性物质,可以在水溶液中自组装成具有疏水核和糖链亲水壳的纳米结构 [26, 27]。同时,CHP纳米颗粒可以吸附Aβ蛋白,防止其沉积和聚集,起到协同作用。作为纳米载体,CHP具有显着优势。
基于以上认识,研究团队在前期设计了DZP-CHP纳米制剂,并确定了药物与纳米载体的最佳给药比例。然后,聚山梨醇酯被吸附在纳米颗粒的表面,以主动靶向通过 BBB 以实现大脑富集。本研究对一系列 DZP-CHP 纳米溶液进行了表征,探索和研究了体外药物释放过程。纳米粒子制备成功后,利用Aβ25-35诱导神经细胞损伤,建立AD细胞模型[28, 29]。然后,研究了DZP-CHP纳米溶液在PC12和SH-SY5Y细胞模型中的保护作用。
材料和方法
材料
使用了以下物质:胆固醇疏水改性支链淀粉(自制)[30];多奈哌齐(上海紫琪生物科技有限公司);聚山梨醇酯80(天津富臣试剂研究所);吲哚菁绿(ICG)染料(天津百盈生物科技有限公司);聚山梨醇酯80(吐温80,PS)(天津富臣试剂办公室);黑鼠(湖南斯莱克精达实验动物有限公司); Aβ25-35(美国西格玛);四甲基偶氮唑盐 (MTT) (US Sigma);新生牛血清(美国 Gibco);乳酸脱氢酶试剂盒(LDH)(南京建成生物有限公司); AO/EB双染荧光试剂盒(中药集团化学试剂有限公司); PC12细胞(大鼠肾上腺嗜铬细胞瘤细胞)购自中南大学湘雅二医院神经内科。 SH-SY5Y细胞(人骨髓神经母细胞瘤细胞)购自ATCC细胞库(美国弗吉尼亚州马纳萨斯)。
纳米粒子的制备
根据文献 [31] 中报道的方法,首先通过水透析成功制备了具有不同 DZP 和 CHP 比率 (w/w)(10:20、4:20 和 2:20)的三种类型的纳米粒子。将一定浓度的 DZP-CHP 纳米颗粒加入到 10 mL 恒定体积的烧杯中,然后吸入另一个装有聚山梨醇酯 80 (PS) 乳化剂(浓度 0.7 mmol)的烧杯中静置 1 小时。然后将混合物置于 EP 管中并超声处理 3 分钟(输出功率 100 W,间歇脉冲工作模式:脉冲宽度 2.0 秒,间歇时间 2.0 秒)。该操作重复 3 次,直到获得均匀的分散体 [32]。过滤去除杂质后,最终得到聚山梨醇酯80乳化多奈哌齐载药纳米粒(PS-DZP-CHP)。
纳米粒子的表征
纳米粒子形态
使用 Tecnai F20 透射电子显微镜分析 DZP 与 CHP 比率为 1:2、1:5 和 1:10 的 DZP-CHP 纳米粒子 (DCP) 的形状、表面形态和尺寸。将一滴 CHP、DZP-CHP 和 PS-DZP-CHP 纳米颗粒置于碳涂层铜网上以形成薄液膜。然后,在薄膜自然干燥后,使用 2% (w/v) 磷钨酸溶液对样品进行负染色。将新鲜制备的纳米颗粒水溶液滴加到干净的硅片上,室温干燥,然后置于JSM-6700F场发射扫描电子显微镜下观察表面结构。
纳米粒子大小和 Zeta 电位
使用动态光散射 (DLS) 分析 DZP-CHP 和 PS-DZP-CHP 纳米颗粒的尺寸、多分散系数 (PDI) 和 zeta 电位。所得均质悬浮液的平均粒径和粒径分布各测定3次。
体外药物释放
使用动态水透析测量多奈哌齐的释放。将 1 毫克 DZP-CHP 和 PS-DZP-CHP 纳米颗粒溶解在 5 毫升磷酸盐缓冲盐水(PBS,pH 7.4,浓度 0.01 M)中,然后转移到透析袋中,透析袋保持在相同的溶液中37°C 恒温,磁力搅拌。在 0、0.5、1、2、4、8、12、24、48 和 72 小时的四毫升 PBS 用相同体积的相同 pH 的 PBS 稀释。紫外-可见分光光度法用于检测透析液在不同时间在 312 nm 处的吸光度;溶液含量用标准曲线测定,体外释放试验重复3次。多奈哌齐的释放百分比按下式计算:
$$Q\% ={{(C{\text{n}} \times V + V{\text{n}}\sum\nolimits_{{{\text{t}} ={0}}}^{ {\text{n}}} {{\text{Ci}}} )} \mathord{\left/ {\vphantom {{(C{\text{n}} \times V + V{\text{n} }\sum\nolimits_{{{\text{t}} ={0}}}^{{\text{n}}} {{\text{Ci}}} )} {(WNP \times LC\% } }} \right. \kern-\nulldelimiterspace} {(WNP \times LC\% }})$$Cn 为 Tn 时间点的样品浓度,μg/mL; V 为 PBS 释放溶液的总体积,mL; Vn 为 Ti 时间点的 PBS 释放液体积,mL; Ci为Ti时间点的多奈哌齐浓度,μg/mL。
等温滴定量热法 (ITC)
将一定浓度的 PS 溶液滴到 CHP 纳米颗粒溶液上,并用 ITC(vip-itc,Microcal,Northampton,MA,USA)测量热量的变化。 CHP 纳米粒子溶液包括三种类型的纳米粒子,它们具有不同的 DZP 与 CHP 的比率(1:2、1:5 和 1:10)。所有溶液在滴定前都经过脱气处理。整个系统的温度保持恒定在 25°C。
观察大脑靶向的动物实验
ICG 标记的多奈哌齐 CHP 纳米颗粒的制备
用分析天平称取400毫克CHP-DZP和20毫克ICG,加入适量DMSO充分混合溶解。然后用移液管将上述得到的溶液滴加到透析袋中,每小时更换一次蒸馏水。三小时后,每2小时更换一次蒸馏水,每次加入400-800毫升蒸馏水,持续48小时,直至DMSO完全透析。之后,用移液管将上述溶液转移到容量瓶中以达到恒定体积,然后用超声波处理2分钟。通过 0.45 μm 滤膜过滤得到 ICG 标记的 DZP-CHP 纳米粒子(ICG-DZP-CHP),将其单独包装并储存在 4°C 的冰箱中以备将来使用。
乳化荧光多奈哌齐 CHP 纳米颗粒的制备
将适量的ICG-DZP-CHP放入10mL烧杯中,加入1‰(v/v)聚山梨醇酯80(PS)乳化剂。将烧杯保持 1 小时,然后转移到 EP 管中,以 100 W 超声处理 2 分钟。重复上述操作 3 次,直至获得均匀的纳米溶液。最终得到过滤并ICG标记的乳化多奈哌齐载药纳米粒(PS-ICG-DZP-CHP)。
验证 APOE 与纳米颗粒结合的 MST 实验
所有 MST 实验均在 Monolith NT.115 系统 (201810-BR-N024) 上进行。所有溶液均用去离子水和分析级试剂制备。缓冲液在室温下制备和储存。蛋白质样品保存在冰上直至使用 [33]。 PS-ICG-CHP 纳米粒子 (55.6 μM) 用去离子水稀释至 40 nM,并加载 ICG 用于荧光。制备APOE溶液(30 μl,55.6 μM),16根毛细管标记为1~16;首先,将 20 μl APOE 加入管 1,然后将 10 μl 加入管 2 至 16。然后,将 10 μL 溶液从管 1 转移到管 2 并充分混合。之后,从管2中取出10μl溶液并转移到管3中。重复该操作直到最终从管16中取出10μl溶液以确保每管中的溶液体积相同。将 10 微升稀释的纳米颗粒添加到每个管中并充分混合以开始测量。使用NT-分析软件对MST测试数据进行分析,并按照软件说明按照质量作用定律进行KD拟合。
用于脑靶向观察的体内荧光成像技术
选择一批每只重约 18-22 克的健康黑色小鼠,随机分为 2 组:PS-ICG-DZP-CHP 组和 ICG-DZP-CHP 组。每只小鼠经尾静脉注射上述组药物200μl 200μg/ml,0.5小时后用1%戊巴比妥钠(50mg/kg)麻醉小鼠。之后,将所有小鼠置于活体成像仪的拍摄区域,成像参数设置为激发波长765 nm-815 nm,吸收波长815 nm-845 nm,获得全动物荧光图像.成像后,解剖所有小鼠,取出肾脏、心脏、脾脏、肺、肝脏和大脑以获得荧光图像。成像参数与上述一致。
纳米粒子的组织分布研究
小鼠分组和采样
45只C57BL/6小鼠随机分为15组:5组注射游离多奈哌齐(游离组),5组注射多奈哌齐纳米粒(纳米组),另外5只注射PS修饰的多奈哌齐纳米粒(PS组) 0.25 mg/kg 通过静脉尾。然后,在注射后 1 小时、3 小时和 6 小时采血。之后,处死所有动物,收集并切碎心、脑、肝和肾组织。然后,精确称量 0.2 g 上述组织并加入约 1 ml 0.9% NaCl 溶液中,并用均质器(65 Hz,150 s)均质。 100 微升组织匀浆准确吸入 1.5 mL EP 管中,加入 0.7 mL 甲醇,涡旋混匀 30 s 使蛋白质沉淀,然后以 12,000 r·min -1 离心 10 分钟。最后取100 μL上清液转移至进样瓶中进行分析。
判断方法
最早使用HPLC进行检测,但灵敏度不够高。因此,进行了后续的 LC-MS 实验,结果显示出很强的特异性,并且没有内源性物质干扰药物测定。 LC-MS 方案符合生物样品测定指南。色谱条件为:流动相A,水(含0.1%甲酸);流动相B,甲醇(含0.1%甲酸);等度洗脱:A30%-B70%;流速,0.3 mL·min −1 ;柱温,35°C;进样量,10 μL。碰撞条件如下:电喷雾电离源 (ESI) 温度,150°C;荒凉气体流量,550 L·h -1 ;荒凉气体温度,500°C。正离子检测条件如下:毛细管电压,3 kV;锥电压,30 V;扫描模式,多反应监测(MRM)。
细胞实验
细胞培养和传代
PC12 和 SH-SY5Y 细胞在添加 10% (v/v) 热灭活胎牛血清 (FBS) 和 1% (v/v) 青霉素和链霉素的高糖 DMEM 中培养,然后保存在含有 5 37°C 时的二氧化碳百分比。细胞在达到 80% 汇合后立即用于各种实验或传代。实验前,根据实验规模所需的细胞密度,将PC12和SH-SY5Y细胞接种于I型胶原蛋白预包被板上。
细胞冷冻保存
显微镜下观察生长至对数期时,将PC12和SH-SY5Y细胞冷冻,PBS洗涤2次,胰酶消化形成细胞悬液,置于无菌离心管中离心收集(1000 r × min − 1 ,3 分钟)。然后加入细胞冷冻保存液,将细胞保存在标有细胞名称和日期的试管中。将细胞置于 4°C 的冰箱中 1 小时,-20°C 的 2 小时,-80°C(冷冻)过夜,最后转移到液氮罐中。
检测细胞存活率的MTT方法
使用MTT还原测定法测量细胞活力。简而言之,将PC12和SH-SY5Y细胞以1 × 10 4 的密度接种于96孔板(预涂I型胶原蛋白)中 细胞/毫升,使细胞粘附到每个孔。孵育 24 小时后,将细胞与不同浓度的多奈哌齐 CHP 纳米溶液或游离多奈哌齐溶液预孵育 2 小时。随后,将 Aβ25-35(终浓度 20 μM)添加到每个孔中。处理过的 96 孔板在 37°C 下孵育 24 小时。之后,加入 MTT(50 μL,5 mg/mL)并与处理过的细胞在 37°C 下孵育 4 小时。最后,小心地除去培养基,将甲臜晶体溶解在150 μL DMSO中。使用酶标仪在 490 nm 处获得吸光度。细胞活力以处理组活细胞百分比和未处理对照组活细胞百分比表示。
细胞上清液中 LDH 活性的测定
PC12和SH-SY5Y细胞以2 × 10 5 的密度接种于96孔培养板(预涂I型胶原蛋白) 和 3 × 10 5 细胞/毫升,分别。孵育 24 小时后,将细胞与不同浓度的多奈哌齐 CHP 纳米溶液或游离多奈哌齐溶液预孵育 2 小时。随后,将 Aβ25-35(终浓度 20 μM)添加到每个孔中。 LDH活性根据试剂盒提供的说明书测量。简而言之,收集带有培养基的培养细胞,然后以 3500 rpm 离心。上清液 (50 μL) 与等体积的反应物混合以引发 LDH 反应。使用酶标仪在 450 nm 处获得吸光度,并计算 LDH 活性。
AO/EB 染色法观察细胞凋亡形态
AO/EB 荧光染料用于评估凋亡细胞的特性。 PC12和SH-SY5Y细胞以3 × 10 5 的密度接种于黑色12孔培养板(预涂I型胶原蛋白) 和 4 × 10 5 细胞/孔,分别。孵育 24 小时后,将细胞与不同浓度的多奈哌齐 CHP 纳米溶液或游离多奈哌齐溶液预孵育 2 小时。随后,将 Aβ25-35(终浓度 20 μM)添加到每个孔中。治疗结束后,按照试剂盒进行操作。整个实验过程中避光。最后观察细胞形态。
检测线粒体膜电位的罗丹明 123 染色方法
MMP 是使用罗丹明 123 (Rh123) 荧光染料测量的,这是一种细胞渗透性阳离子染料,由于其高度负性,它优先分布到线粒体中。 PC12和SH-SY5Y细胞以2 × 10 5 的密度接种于黑色24孔培养板(预涂I型胶原蛋白) 和 3 × 10 5 细胞/孔,分别。孵育 24 小时后,将细胞与不同浓度的多奈哌齐 CHP 纳米溶液或游离多奈哌齐溶液预孵育 2 小时。随后,将 Aβ25-35(终浓度 20 μM)添加到每个孔中。处理后,将细胞用 PBS 洗涤并与 10 μg/mL 罗丹明 123 一起在黑暗中在 37°C 下孵育 30 分钟。孵育后,用PBS洗涤细胞3次,用荧光读板仪在488 nm和510 nm处测量荧光强度。
统计处理和数据分析
所有实验均重复3次,结果以均值 ± 标准差表示。使用GraphPad Prism统计软件,单向方差分析,Student's t 检验等方法进行统计分析。 P <0.05 表示差异具有统计学意义。
结果
纳米粒子的特性
CHP 自聚集形成具有疏水核的纳米颗粒,可以装载 DZP。药物与纳米材料的比例为 1:2、1:5 和 1:10 的载有 DZP 的 CHP 纳米粒子 (DCN) 被命名为 DCN1、DCN2 和 DCN3。根据扫描电镜结果,CHP纳米颗粒呈球形结构,加载DZP后,DCNs也呈球形,如图1所示。CHP纳米颗粒的平均尺寸和zeta电位分别为257 ± 3.05 nm和-2.81分别为 ± 0.27 mV。 DZP 加载后,平均大小分别为 273 ± 3.72、260.7 ± 1.76 和 266.8 ± 4.56 nm,zeta 电位分别为 -6.20 ± 0.40、-5.75 DC ± 0.40、-5.75 DC ±3.75 DC,-5.75 DC ±3.56 和 DC ±3.56 和 N。 ,如表1所示。药物包封率分别为42.00 ± 5.65%、86.54 ± 1.31%和59.71 ± 4.43%,载药率分别为12.02 ±4.43%、12.02 ±3.7±3.7±3.7%分别。
<图片>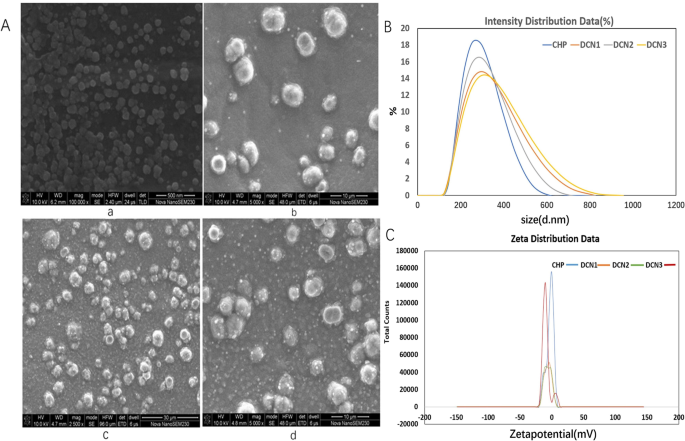
扫描电子显微镜图像 (a , a-CHP, b-DCN1, c-DCN2, d-DCN3), 尺寸分布 (b a-CHP、b-DCN1、c-DCN2、d-DCN3) 和 zeta 电位 (c a-CHP, b-DCN1, c-DCN2, d-DCN3) 纳米颗粒
ITC 衡量
对于 DCNs,在整个反应过程中,反应主要表现为一个向上的峰(图 2),该反应是吸热的,因为向上的峰表明是放热反应。因此,PS 可以自发地吸附在 DCN 表面。 PS亲和力为(14.7 ± 2.76) × 10 4 M −1 , (29.8 ± 1.66) × 10 4 M −1 和 (36.7 ± 3.84) × 10 4 M −1 ,DCN1、DCN2和DCN3的PS覆盖程度分别为2.65 ± 0.193、2.70 ± 0.372和1.49 ± 0.434。该结果表明 PS 以高亲和力吸附到 DCN 表面并且在 DCN2 上具有更大的覆盖量。 ΔH> 0 和 ∆S> 0 表示三个粒子主要通过与 PS 的疏水相互作用结合。
<图片>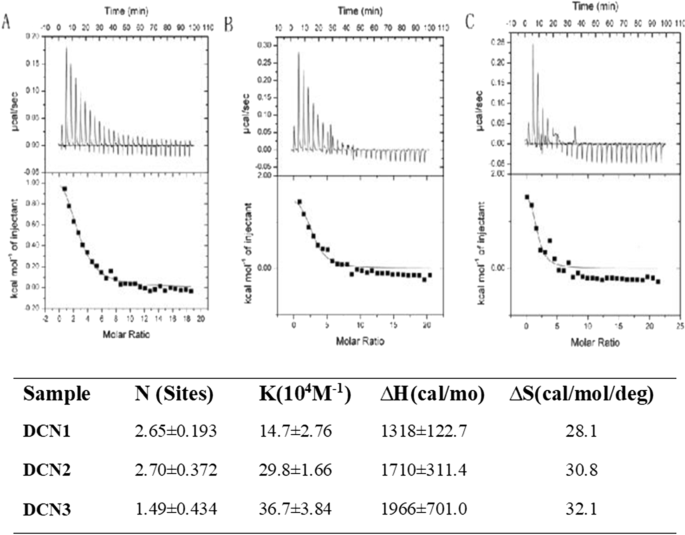
PS (0.9 mM) 滴定到 a 的等温量热数据 DCN1,b DCN2 和 c 25°C 下的 DCN3 (0.02 mM) 溶液。滴定到 NP 溶液后 PS 与纳米颗粒 (NPs) 结合的覆盖度、亲和力 (KA) 以及焓和熵的变化
三种纳米粒子类型的表征
通过透析制备的 CHP NPs 和 DCNs 显示出均匀的球形(图 3a)。根据上述研究,我们选择药物与纳米材料比为1:5的DZP-CHP纳米粒作为以下实验的对象。 DZP-CHP纳米颗粒具有相对均匀的粒径260.7 ± 1.76 nm,分散指数为0.196 ± 0.019。尽管载药后粒径保持相对稳定,但在 PS 吸附后粒径急剧增加至 335.2 ± 5.46 nm。 DZP-CHP纳米颗粒的zeta电位为-0.66 ± 0.04 mV,用聚山梨醇酯80包覆后,zeta电位降至- 2.22 ± 0.86 mV(图3b)。
<图片>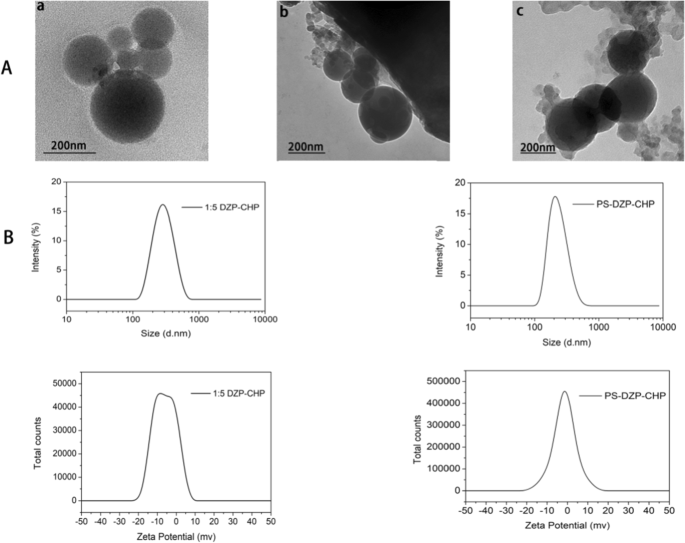
不同纳米颗粒的表征。 一 a CHP NPs 的透射电子显微镜照片,b DZP-CHP NPs 的透射电子显微镜照片,c PS-DZP-CHP NPs 的透射电子显微镜照片。 B:CHP-DZP NPs(进料比1:5)和聚山梨醇酯80改性DZP-CHP NPs(进料比1:5)的粒径图和zeta电位图
纳米颗粒的体外药物释放
结果表明,与游离多奈哌齐相比,DZP-CHP NPs和PS-DZP-CHP NPs释放DZP 72 h,控释效果明显。载药纳米粒的早期快速释放可能是由于药物分子的快速溶解和释放,然后,缓释可能是由于药物浓度的降低引起的,而后者只能受溶解和扩散的影响。然后研究了用聚山梨醇酯 80 包被和未包被的 DZP-CHP NPs 的体外药物释放。 PS-DZP-CHP NPs释放较慢的原因可能是聚山梨醇酯80对疏水性小分子药物有很强的吸附作用(图4)。
<图片>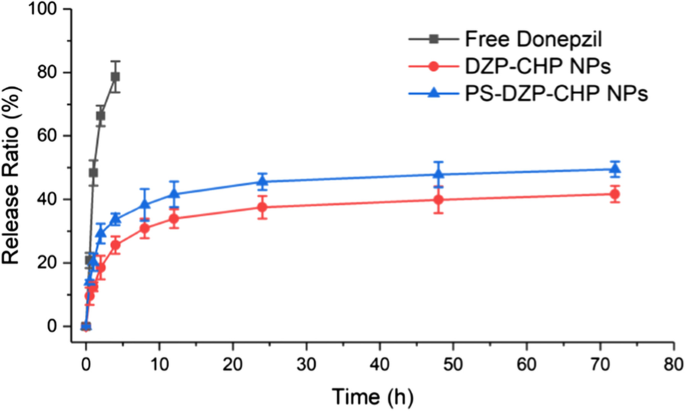
DZP、DZP-CHP NPs (1:5)和PS-DZP-CHP NPs的体外药物释放曲线
纳米粒子脑靶向效应
使用实时荧光成像技术观察大脑靶向
注射游离 ICG 的小鼠大脑没有显示荧光,但注射 PS 乳化的纳米颗粒的大脑比注射非乳化纳米颗粒的大脑显示出更强的荧光,因为两种纳米颗粒在通过尾静脉注射后都能到达大脑(图. 5a)为了验证这一点,我们在静脉注射 ICG-DZP-CHP 和 PS-ICG-DZP-CHP 溶液后 30 分钟解剖小鼠,取出研究所需的所有器官,然后进行荧光成像。 PS-ICG-DZP-CHP 纳米粒子在大脑中呈现出强烈的荧光,但在其他器官中没有观察到(图 5b)。图像显示,经 PS 修饰的纳米粒子在脑组织中呈现出最强的荧光,而未经修饰的纳米粒子则呈现出较弱的荧光。注射游离ICG的小鼠脑组织未观察到荧光(图5c)。
<图片>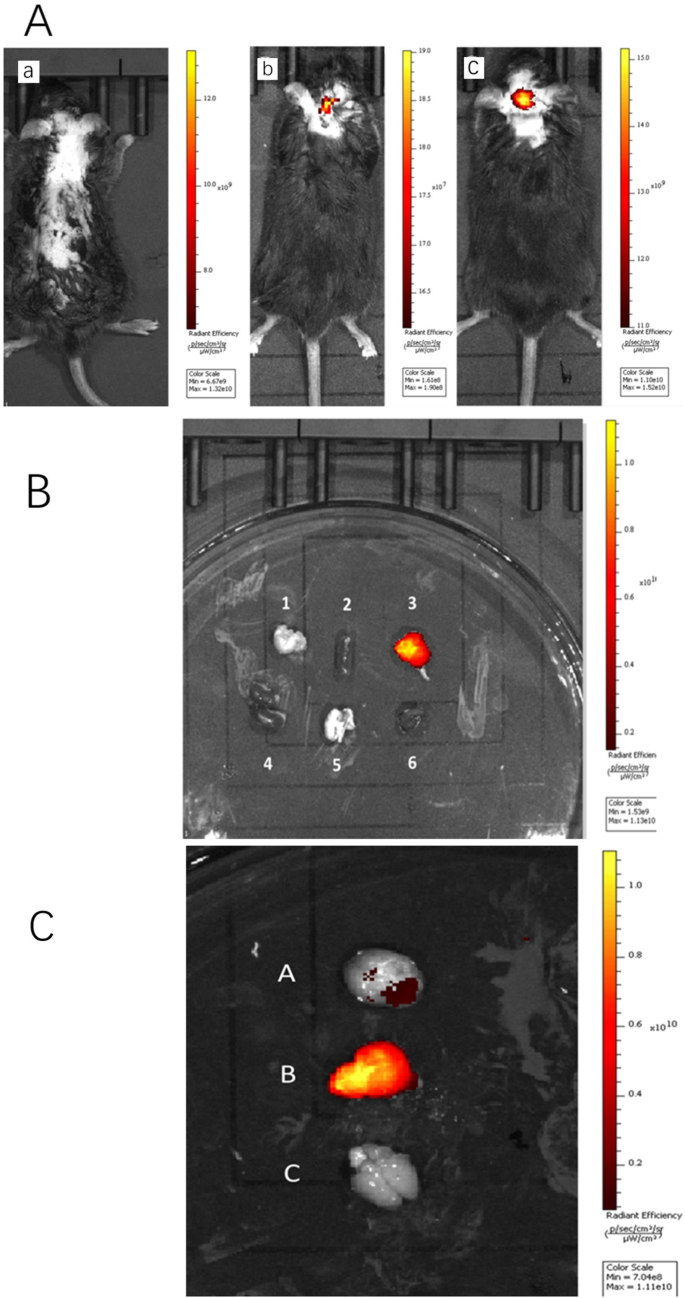
通过尾静脉注射不同溶液后的体内荧光图像。 一 整个动物的荧光图像通过尾静脉注射了 200 μg/ml DZP-CHP 或 PS-DZP-CHP 纳米粒子,其中载有 ICG 作为染色剂。 a Free ICG 溶液(荧光强度 × 10 9 ), b ICG-DZP-CHP纳米粒子(荧光强度 × 10 7 ), c PS修饰的ICG-DZP-CHP纳米粒子(荧光强度 × 109)。 b Fluorescence images of various organs after injection of DZP-CHP nanoparticles modified by PS via the tail vein. c Fluorescence images of the brain after dissection. d Brain of mice injected with ICG-PS-DZP-CHP nanoparticles, e brain of mice injected with ICG-DZP-CHP nanoparticles modified with PS, f brain of mice injected with free ICG
Tissue Distribution of Nanoparticles in Mice
Donepezil was distributed in various tissues and mainly in the brain after injection of PS-DZP-CHP nanoparticles. Because it is metabolized through the kidney, the concentration of donepezil is very high in the kidney at certain times (Fig. 6a). In the brain, the concentration of free donepezil reached a peak in a very short time and then decreased rapidly. However, the concentration of donepezil nanoparticles reached a peak much more slowly and then decreased, especially nanoparticles modified with PS, which indicates a sustained-release effect with a delayed peak and prolonged retention time. Obviously, the nanoparticles improved the bioavailability of the drug (Fig. 6b).

一 The concentration of donepezil in the brain, heart, liver and kidney at different times. b The concentrations of free DZP, DZP-CHP nanosolution and DZP-CHP nanosolution modified with PS in brain tissue at different times
MST Results
MST results showed that the thermal surge changed regularly with increasing ligand concentration, and the KD value was 3.63 μM, indicating that the ligand effectively binds to the target protein, which verifies that PS can bind to Apo E and is relatively stable. After surface modification with PS, CHP nanoparticles can promote adsorption of Apo E, theoretically confirming that the nanoparticles we designed can specifically target brain tissue because the nanoparticles adsorbed Apo E, which may mediate passage through the blood–brain barrier (Fig. 7).

一 Raw MST data. The fluorescently labeled molecules were observed for 5 s. At this time, the infrared laser was turned on, and a small part of the capillary tube was heated to 2–5 °C. The molecules migrated along a thermal gradient, resulting in changes in fluorescence intensity. When the infrared laser is turned off, the molecules diffuse along the concentration gradient. b The binding curve is generated by the difference between the initial fluorescence intensity and the intensity in the presence of heat, and the curve conforms to the standard 1:1 binding model
Establishment of a Nerve Injury Model Induced by Aβ25–35
MTT tests were used to detect the effect of different concentrations of Aβ25-35 on the activity of PC12 and SH-SY5Y cells [Fig. 8a (i), Fig. 8b (i)], and the results showed that with increasing Aβ25–35 concentration, PC12 and SH-SY5Y cell proliferation activity gradually decreased compared with that in the normal control group. When PC12 and SH-SY5Y cells were treated with 20 μM Aβ25–35, PC12 cell activity decreased to 49.5 ± 3.3% that observed in the control group (P < 0.01), and SH-SY5Y cell activity decreased to 49.7 ± 0.8% (P < 0.01). An LDH kit was used to detect the effect of different concentrations of Aβ25–35 on LDH activity in both cell supernatants. Colorimetric tests showed that activity in both supernatants increased gradually with increasing Aβ25–35 concentration. After treatment of the cells with 20 μM Aβ25–35, PC12 cell LDH release increased to 359.3 ± 18.3% that in the control group (P < 0.01), and SH-SY5Y cell LDH release increased to 360.0 ± 18.2% (P < 0.01). Rhodamine 123 staining was used to detect the effect of different concentrations of Aβ25– 35 on the mitochondrial membrane potential in both cell lines [Fig. 8a (ii), b (ii)]. The test showed that the mitochondrial membrane potential in both cell lines decreased gradually with increasing Aβ25– 35 concentration (5, 10, 20, 40 μmol/L). Treatment of the cells with 20 μM Aβ25–35 decreased the PC12 cell mitochondrial membrane potential to 51.3 ± 1.6% that in the control group (P < 0.01); for SH-SY5Y cells, the MMP decreased to 47.9 ± 1.7% that in the control group (P < 0.01).

Effects of different concentrations of Aβ25–35 on injury to PC12 and SH-SY5Y cells. 一 , b The effect of different concentrations of Aβ25–35 on the cell survival rate, LDH activity and cell mitochondrial membrane potential in PC12 cells and SH-SY5Y cells (*P < 0.05, ** P < 0.01 vs control group). c The effect of different concentrations of Aβ25– 35 on damage to the morphology of PC12 cells:a control group, b Aβ25– 35 (5 μM) injury group, c Aβ25– 35 (10 μM) injury group, d Aβ25– 35 (20 μM) injury group, e Aβ25– 35 (40 μM) injury group. D:the effect of different concentrations of Aβ25– 35 on injury to the morphology of SH-SY5Y cells:f—control group, g Aβ25– 35 (5 μM) injury group, h—Aβ25– 35 (10 μM) injury group, i—Aβ25– 35 (20 μM) injury group, j—Aβ25– 35 (40 μM) injury group
Inverted fluorescence microscopy was used to observe morphological changes of PC12 and SH-SY5Y cells injured by different concentrations of Aβ25– 35 (Fig. 8c, d). The PC12 and SH-SY5Y cells in the control group had higher density, fusiform shapes, fuller cell bodies and longer protrusions. As the concentration of Aβ25– 35 increased (5, 10, 20, 40 μmol/L), the number of cells in both cell lines gradually decreased, the cell bodies shrank slightly, and the protrusions began to shrink sharply. When the concentration of Aβ25– 35 was increased to 40 μmol/L, the protrusions broke significantly, most of the cells contracted sharply, their shape became irregular, and some cells detached and became suspended in the solution.
Therefore, we treated PC12 and SH-SY5Y cells with 20 μM Aβ25-35 for 24 h to establish a nerve injury model.
Neuroprotective Effect of Drug-Loaded Nanoparticles (DZP-CHP)
MTT assays were used to detect the activity of different concentrations of DZP and DZP-CHP (2.5 μM, 5 μM, 10 μM) in PC12 and SH-SY5Y cells [Fig. 9a (i), b(i)]. Tests showed that treatment of PC12 cells with 20 μM Aβ25-35 alone resulted in a significant reduction in cell viability to 48.4 ± 2.8% that in the control group (P < 0.01). However, after pretreatment with DZP and DZP-CHP (2.5 μM, 5 μM, 10 μM) solutions, the viability of PC12 cells increased significantly. The viability of PC12 cells in the DZP-CHP group was higher than that in the DZP group (P <0.05)。 Similarly, treatment of SH-SY5Y cells with 20 μM Aβ25– 35 alone resulted in a significant reduction in cell viability to 48.5 ± 4.0% that in the control group (P < 0.01), while after pretreatment with DZP and DZP-CHP solution (2.5 μM, 5 μM, 10 μM, respectively), the viability of SH-SY5Y cells increased significantly. The viability of SH-SY5Y cells in the DZP-CHP group was higher than that in the DZP group (P <0.05).

Effect of DZP-CHP on Aβ25-35-injured PC12 and SH-SY5Y cells. 一 and b The effects of DZP-CHP on the cell survival rate, LDH activity, and mitochondrial membrane potential of PC12 and SH-SY5Y cells injured by Aβ25-35 (#P < 0.05, ## P < 0.01 vs Aβ25-35 group; ▲ P < 0.05 vs DZP group). c The effect of DZP-CHP on the apoptosis morphology of PC12 cells injured by Aβ25-35; a—control group, b—Aβ25-35 injury group, c—DZP (5 µM), d—DZP -CHP (5 µM), e—DZP (10 µM), f—DZP-CHP (10 µM). D:the effect of DZP-CHP on the apoptosis morphology of SH-SY5Y cells injured by Aβ25-35; g—control group, h—Aβ25-35 injury group, i—DZP (5 µM), j—DZP-CHP (5 µM), k—DZP (10 µM), l—DZP-CHP (10 µM). E:red/green fluorescence ratio
LDH kits were used to detect the effects of different concentrations of DZP and DZP-CHP (5 μM and 10 μM) on the release of LDH from PC12 and SH-SY5Y cells into the culture medium [Fig. 9a (ii), b (ii)]. Colorimetric measurements showed that the release of LDH from PC12 cells that were exposed to 20 μM Aβ25-35 alone increased significantly by 355.1 ± 16.6% (P < 0.01). In the presence of DZP and DZP-CHP (5 μM and 10 μM), LDH release from PC12 cells dropped significantly. The effect in the DZP-CHP group was higher than that in the DZP group (P < 0.01). Similarly, the release of LDH from SH-SY5Y cells exposed to 20 μM Aβ25-35 increased significantly to 357.8 ± 12.5% (P < 0.01). However, after pretreatment with DZP and DZP-CHP (5 μM and 10 μM), LDH release from SH-SY5Y cells decreased significantly. The effect in the DZP-CHP group was higher than that in the DZP group (P <0.05).
According to previous reports, depolarization of the MMP leads to loss of Rh123 from mitochondria, which in turn leads to a decline in intracellular fluorescence. Therefore, to characterize changes in the mitochondrial membrane potential in PC12 and SH-SY5Y cells treated with Aβ25-35, DZP, and DZP-CHP (5 μM, 10 μM), rhodamine 123 was used for detection [Fig. 9a (iii), b (iii)]. The results showed that the fluorescence intensity of rhodamine 123 decreased significantly to 44.3 ± 3.8% (P < 0.01) after incubation of PC12 cells with 20 μM Aβ25-35 for 24 h. However, pretreatment with DZP and DZP-CHP (5 μM, 10 μM) solutions resulted in a significant increase in fluorescence intensity in a dose-dependent manner, and the effect in the DZP-CHP group was higher than that in the DZP group (P <0.05)。 Similarly, after treatment of SH-SY5Y cells with 20 μM Aβ25-35 for 24 h, the fluorescence intensity of rhodamine 123 significantly decreased to 42.5 ± 4.6% (P < 0.01). However, after pretreatment with DZP and DZP-CHP (5 μM, 10 μM), the fluorescence intensity increased significantly, and the effect in the DZP-CHP group was higher than that in the DZP group.
An AO-EB double staining kit was used to detect morphological changes of PC12 and SH-SY5Y cells treated with different concentrations of DZP and DZP-CHP (5 μM and 10 μM) (Fig. 9c, d). After AO-EB double staining, the nuclei of living cells presented green fluorescence under a fluorescence, and the fluorescence of apoptotic cells was orange-red; the higher the degree of apoptosis is, the brighter the fluorescence. Compared with the untreated control group, PC12 and SH-SY5Y cells treated with Aβ25-35 alone showed typical apoptotic characteristics, such as highly condensed and broken nuclei and obvious cell injury. However, pretreatment with DZP and DZP-CHP solution (5 μM and 10 μM) significantly inhibited cell damage and improved cell morphology.
Discussion
Although nanoparticles have been shown to be an effective delivery medium for nervous system diseases, the complexity of their structure and performance makes it challenging to detect and evaluate their physical–chemical properties and biological safety [34,35,36]. Therefore, after nanodrugs are designed, experiments to verify the safety and effectiveness of the nanomaterials at the cell and animal levels are extremely necessary. In the early laboratory stage, PS-DZP-CHP nanoparticles were successfully synthesized, and the optimal dosing ratio of the drug to CHP was set at 1:5. Due to the stable adsorption of PS to apolipoproteins ApoB and ApoE, brain targeting of nanoparticles can be achieved by permeation through the BBB [37]. The expected goal is that nanoparticles begin to decompose after reaching the brain; DZP is released and increases the concentration of cholinesterase; CHP reduces Aβ protein deposition and improves the brain environment; and administration frequency decreases because of the sustained release from nanoparticles [38,39,40,41].
Therefore, this study conducted an in vitro drug release test to assess the sustained release effect of nanoparticles. Compared with free DZP, nanoparticles achieved local sustained release in the brain, and PS can adsorb plasma proteins and reduce the loss of nanoparticles and prolong the release time to achieve a long cycle. After injection of ICG-labeled DZP-CHP and PS-DZP-CHP nanoparticles into rats, emulsified nanoparticles showed stronger fluorescence in the brain than those not emulsified with Tween 80. After organ biopsy, fluorescence imaging of various organs revealed that only the brain presented strong fluorescence, while other organs did not, indicating that nanoparticles did not release the drug until they reached the brain, which met the expected goal. In addition, nanoparticles modified with polysorbate 80 adsorbed ApoE to the surface and simulated low-density lipoprotein to bind to lipoprotein receptors on the surface of endothelial cells and enter the brain through LDLR induction [42, 43]. Moreover, the mechanisms by which nanoparticles regulate the tight junctions between endothelial cells and the inhibition of P-glycoprotein may produce a synergistic effect on their transcellular transport into the brain parenchyma [44]. However, since polysorbate 80 is prone to produce toxic substances, which cause untoward reactions, such as hypotension, dyspnea and shock, the dosage should be strictly controlled [45, 46]. Although a large number of nanodrugs have been developed for treatment of CNS diseases, most have shown poor effects [4]. In this study, we built a brain model of AD patients to evaluate the protective effect of nanoparticles on the brain. Because the toxicity of the Aβ protein to nerve cells varies with the concentration, it is better to screen an appropriate concentration to better simulate the brain environment of AD patients. The model was treated with DZP and DZP-CHP nanosolutions in advance to investigate the protective effect of DZP-CHP against PC12 and SH-SY5Y cell damage induced by Aβ25-35. The results showed that both solutions improved the cell proliferation activity caused by Aβ25-35, LDH release declined, and the mitochondrial potential rose. The inhibitory effect of the DZP-CHP nanosolution on cell damage induced by Aβ25-35 was significantly better than that of free DZP solution, and a concentration of 10 M DZP-CHP nanosolution proved to be the best, which may be due to the optimal drug concentration approach.
Basically, this study deduced the activity process of nanoparticles in vivo , verifying that PS-DZP-CHP nanoparticles have strong brain targeting, a good sustained release effect and a good AD therapeutic effect, thus demonstrating that they are clinically promising nanodrugs. The difficulty of treating the brain with medication has always been a major problem for researchers. The BBB leads to difficulty in curing CNS diseases, such as Parkinson's disease, brain tumors and brain stroke [47]. PS-DZP-CHP nanoparticles can effectively pass the BBB without destroying it, and DZP can be replaced as a model drug. Loading other drugs with high fat solubility and poor dissolution in vivo into the hydrophobic center of the NPs can not only increase brain targeting but also improve the water solubility of the drug. In addition, the design of CHP nanoparticle solutions is simple, and the drug dosage of the hydrophobic center can be flexibly controlled to ensure the best therapeutic effect while minimizing cytotoxicity [48, 49]. In future work, we will conduct in vivo research on DZP-CHP nanoparticles, such as evaluation of drug metabolism and side effects. These studies supplied a new strategy for brain drug delivery, and it is advantageous to clinical application of nanodrug with AD treatment.
Conclusion
DZP-CHP nanoparticles showed an optimal drug to nanomaterials dosing ratio of 1:5, which led to higher PS coverage and drug loading. DZP-CHP nanoparticles with PS adsorption exhibited slow release and significant brain targeting. Nanoparticle surface modification with PS can promote adsorption of Apo E and thus is vital for brain targeting. DZP-CHP nanoparticles had a protective effect on neurotoxicity and were superior to free donepezil.
数据和材料的可用性
不适用。
纳米材料
- IBM 科学家率先演示用于纳米粒子的摇摆布朗马达
- 用于化学传感器的金纳米粒子
- 用于改进诊断和治疗应用的多功能金纳米粒子:综述
- 用于癌症治疗的纳米粒子:当前的进展和挑战
- 斑马鱼:一种用于纳米技术介导的神经特异性药物递送的有前景的实时模型系统
- 用于神经保护的脑靶向聚山梨醇酯 80 乳化多奈哌齐载药纳米颗粒
- 用贵金属纳米粒子装饰的电纺聚合物纳米纤维用于化学传感
- 含有用于甘油电渗析脱盐的无机离子交换剂纳米颗粒的复合膜
- 用于体内 CT 成像和肾脏清除特性的新型生物相容性 Au Nanostars@PEG 纳米颗粒
- 用于细胞内蛋白质递送的二氧化硅纳米颗粒:一种使用绿色荧光蛋白的新型合成方法
- 合成单分散二元 FePt-Fe3O4 纳米粒子的后处理方法
- 神经退行性疾病的计算机模拟