

探测 Ag n V (n =1-12) 簇的结构、电子和磁特性
摘要
Ag n 的结构、电子和磁性特性 V (n =1-12) 簇已经使用密度泛函理论和 CALYPSO 结构搜索方法进行了研究。几何优化表明,低能 AgnV 簇中的钒原子有利于最高度协调的位置。 Ag n 中一个 V 原子替换一个 Ag 原子 + 1 (n ≥ 5) 集群修改了宿主集群的最低能量结构。 Ag n的红外光谱、拉曼光谱和光电子光谱 V (n =1-12) 簇被模拟,可用于确定未来最稳定的结构。通过原子平均结合能、解离能和能隙分析基态的相对稳定性、解离通道和化学活性。发现V原子可以提高宿主簇的稳定性,Ag2除外。最可能的解离通道是 Ag n V =Ag + Ag n - 1V for n =1 和 4-12 和 Ag n V =Ag2 + Ag n - 2V for n =2 和 3. Ag n 的能隙 具有奇数 n 的 V 簇 远小于 Ag n + 1 个集群。磁性能分析表明,Ag n 的总磁矩 V簇主要来自V原子,从1到5μ不等 B. V和Ag原子之间的电荷转移应负责磁矩的变化。
背景
在过去的几十年里,银团簇因其异常的光学和催化特性而受到特别关注 [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15, 16、17、18、19、20]。同时,理论和实验研究表明,一个原子掺杂到另一个元素的小簇中可以从根本上改变宿主簇的性质 [21,22,23,24,25,26,27,28,29,30,31 ,32,33,34,35,36,37,38,39,40,41,42,43,44]。掺杂不同原子的银簇有望为成像、传感、生物学、医学和纳米技术的潜在应用量身定制所需的光学、电子和磁特性 [45,46,47,48,49,50,51,52 ,53,54,55]。例如,Si 掺杂到银团簇中会导致银团簇的紫外-可见吸收光谱峰变宽和衰减[45]。 Ag n的光学特性 金米 可以通过改变银原子与金原子的比例来调整,Au4Ag4 可能是一种有潜力的分子光电器件 [46]。与银簇相比,二元 Ag-Au 簇修饰的 TiO2 电极提高了太阳能电池的短路电流密度和最大功率转换效率 [47]。一组典型配体(-COOH、-CN、-OH、-SH、-CH3、-NO2、-NH3、-NO)的吸附能在 Ag12Au 簇上比在 Ag13 簇上小 [48]。 Ag-Cu 纳米合金是替代碱性燃料电池中贵重 Pt 基催化剂的潜在候选者 [49]。 Ag12Cu 簇的外层原子中的电子比 Ag13 簇具有更活跃的特性[50]。 Ag-Pd 合金簇对氢解离的催化活性与化学计量密切相关。 Ag6Pd2 是最有效的氢分子吸附簇,可以作为 H2 存储的有希望的候选者 [51]。单个 3d 过渡金属原子的引入有效地解决了 Ag12 二十面体的不稳定性问题 [52]。最近,由于其独特的物理和化学性质,对 V 掺杂的银团簇进行了一些研究 [56,57,58,59]。张等人。据报道,与纯二十面体 Ag13 簇相比,中性 Ag12V 簇显示出更大的相对结合能 [56]。陈等人。发现 V@Ag12 上的吡啶 − 簇表现出最强的化学增强,因子约为一千 [57]。梅德尔等人。探索了 Ag n 中价态跃迁和自旋矩的性质 V + 对 n 具有增强稳定性的集群 =14 [58]。然而,关于中性 V 掺杂的银团簇的工作相对较少。特别是 Ag n 的各种光谱 V簇尚未获得,但对簇结构的识别非常有帮助。还需要进一步探索 V 掺杂银簇的结构基序。嵌入非磁性主体中的磁性杂质的磁矩变化仍未完全了解。因此,在本文中,Ag n 的几何、电子和磁特性 V (n =1-12) 簇将通过密度泛函理论 (DFT) 进行系统研究。希望该工作能为理解材料的功能与结构的关系及相关实验提供参考。
方法
在 GAUSSIAN09 程序包(Frisch, M. J. et al., Wallingford, KY, USA)[60] 中实现的不同交换相关泛函的准确性首先通过对 Ag2 二聚体的计算得到验证。基于 PW91PW91/LanL2DZ (Perdew, JP et al., New Orleans, Louisiana, USA) 水平的计算结果与实验结果 [61, 62] 非常吻合,如表 1 所示。另一方面,测试计算对 AgV 二聚体使用不同的 DFT 泛函。表 1 中列出的五个功能支持相同的自旋配置。因此,该理论水平用于 Ag n 的几何优化和频率分析 V簇。 Ag n 的许多初始配置 V簇是通过使用CALYPSO构建的,这是一种有效的结构预测方法[63]。在这种方法中,结构演化是通过粒子群优化 (PSO) 实现的,粒子群优化是一种基于群体的随机优化技术。键表征矩阵技术用于提高搜索效率并去除相似结构。 CALYPSO 的显着特征只需要给定簇的化学成分即可预测其结构。由于自旋极化效应,每个初始结构都在可能的自旋状态下进行了优化。如果找到一个虚振动频率,不稳定结构就会松弛,直到真正获得局部最小值。在所有计算中,收敛阈值设置为 6.0 × 10 −5 Å 为位移,1.5 × 10 −5 力的 Hartree/Bohr 和 10 −6 Hartree 获得总能量。
结果和讨论
几何结构和振动光谱
对于 Ag n V (n =1-12) 簇,进行了广泛的结构搜索并获得了许多异构体。每个 Ag n 的最稳定结构和两个低位异构体 V簇如图1所示,按照能量从低到高,这些异构体分别用na、nb、nc表示,其中n 代表Agn中Ag原子的数量 V簇。与每个最稳定的结构相比,它们的对称性、自旋多重性和能量差异也显示在图中。基态Ag n的一些物理参数 V簇聚集在表2中。同时,为了检查掺杂剂V对银簇的影响,Ag n的几何优化 (n =2–13) 聚类已使用相同的方法和基组完成。 Ag n 的最低能量结构 图 1 中绘制的聚类与之前的报告 [39] 一致。
<图片>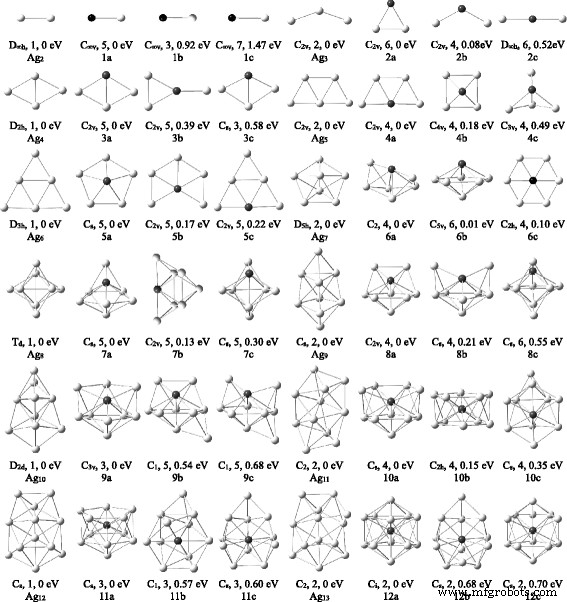
Ag n的基态结构 + 1 和 Ag n V (n =2–12) 簇。 Ag n 的两个低位异构体 V簇。下面给出了对称性、自旋多重性和能量差。灰色和黑色球分别表示Ag和V原子
AgV 二聚体的优化结果表明,五重态自旋态在能量上比三重态和七重态自旋态分别低 0.92 和 1.47 eV。因此,五重奏 AgV 是基态结构。 Ag2V 团簇最稳定的结构是具有 C2v 对称性的三角形 2a。四重旋态的 2a 构型变成 2b 异构体。 3a 和 4a 异构体类似于 Ag4 和 Ag5 簇的最低能量结构,是 Ag3V 和 Ag4V 簇的基态。 Ag4V簇的基态结构也与Medel等人的结果一致。 [58]。顶部带有 V 原子的 4b 异构体是一个方形金字塔和第一个三维 (3D) 结构。 4c 异构体具有三角形双锥结构,其总能量比 4a 异构体高 0.49 eV。其他平面和3D异构体不如4c异构体稳定。
从 n 开始 =5,Agn的最低能量结构 V 群集更喜欢 3D 配置。为了防止遗漏基态,我们还利用了从稳定的银簇中用一个 V 原子取代一个 Ag 或将 Ag 原子添加到小 Ag n 的优化策略 V簇。 5a 和 6a 异构体是 Ag5V 和 Ag6V 簇最稳定的结构。这两种异构体是通过将几何形状从 C5v 和 C2v 分别扭曲为 Cs 和 C2 点群而获得的。 6a 异构体在四重旋转状态下比在六重旋转状态下低 0.62 eV。 5c 和 6b 异构体类似于纯 Ag6 和 Ag7 簇的基态结构。 6b 异构体几乎与 6a 异构体简并。由于Jahn-Teller效应,具有C2h对称性的平面6c异构体与D2h对称性略有偏差。
关于 Ag n V (n =7-12)簇,异构体的数量随着簇大小的增加而迅速增加。优化后的结构表明 Ag n 的能量 具有相同构型的V簇随着V原子配位数的减少而增加。结果,各种 Ag n 进一步考虑 V 原子占据配位数最高位置的 V 异构体,以确保最稳定的结构是全局最小值。 Ag7V、Ag8V、Ag9V、Ag10V、Ag11V 和 Ag12V 团簇的最低能量结构分别为图 1 中的 7a、8a、9a、10a、11a 和 12a。它们的几何形状与 Medel 等人的结果定性一致。 [58]。这些结构与相应的Agn的基态结构完全不同 + 1 个簇并包含一个五边形双锥体。 Ag n 对应于 Ag n 最低能量结构的 V 异构体 + 1 个簇位于每个基态结构 (na) 上方。此外,10b 和 12a 与 D5d 和 D3d 对称性略有偏差。 Ag12V团簇的笼构型,V原子占据中心位置,仅在最低自旋态下被发现。
从优化结果可以看出,Ag n V簇具有明显的生长规律。梯形和二十面体是Ag n生长过程的两个基本框架 V簇,如图2所示。Agn的二维到三维结构转变 V簇出现在n =5. Ag n 的跃迁尺寸 V簇比纯Ag簇小(n =6)。对于 n =5-12,Ag n 的基态 V簇明显不同于Ag n + 1 个集群。 Ag n 中的 V 原子 V簇往往占据最高配位的位置,并逐渐被Ag原子包裹在中心。这可能归因于配合物化学键理论中的最大重叠原理。由于在上述情况下 Ag 和 V 原子的轨道重叠较多,因此 Ag n 的能量 V簇也与Ag原子的排列有关,V簇越低,相应簇越稳定。
<图片>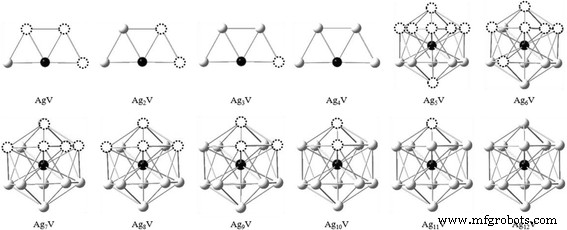
Ag n的生长图 V (n =1-12) 个簇
红外光谱和拉曼光谱是识别团簇结构和材料成分的有力工具。通常,结构识别是通过将实验结果与理论预测进行比较来完成的,这是必不可少的一部分。因此,最稳定的Agn的红外光谱和拉曼光谱 V (n =1-12) 团簇如图 3 所示。红外光谱显示极群不对称振动。拉曼光谱揭示了非极性基团和骨架的对称振动。 AgV 二聚体具有相同的红外和拉曼光谱。对于其他 Ag n V簇,红外光谱的强吸收位置在拉曼散射光谱中有一个弱峰。相反,拉曼散射峰强,红外吸收弱。所有异构体在两种光谱中的峰位置均在15~270 cm −1 .每个 Ag n 的红外光谱中最强烈的峰 V簇与Ag-V伸缩振动有关。
<图片>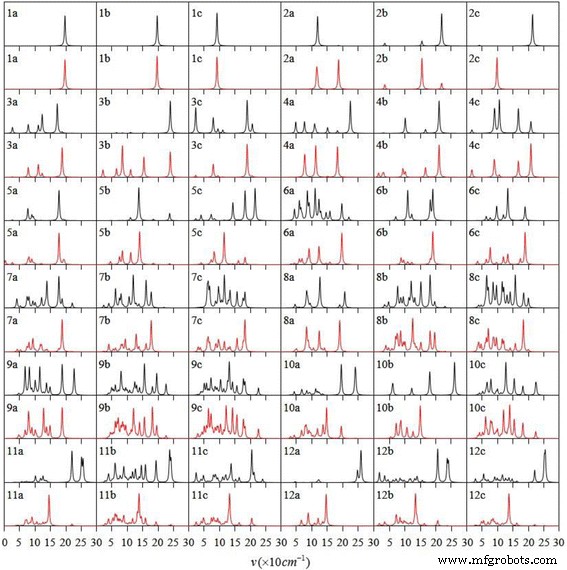
Ag n 基态和两种低位异构体的红外光谱(黑色)和拉曼光谱(红色) V (n =1-12) 个簇
电子属性
垂直电离势 (VIP) 和电子亲和势 (EA) 是探测电子特性的两个主要量,计算如下:
$$ \mathrm{VIP}=E\left(\mathrm{cationic}\ \mathrm{cluster}\right)-E\left(\mathrm{cluster}\right) $$ (1) $$ \mathrm{EA }=E\left(\mathrm{cluster}\right)-E\left(\mathrm{anionic}\ \mathrm{cluster}\right) $$ (2)其中 E (阳离子簇)和 E (阴离子簇)是中性簇几何中阳离子和阴离子簇的单点能量。对于最低能量 Ag n + 1 和 Ag n V 集群,表 3 列出了计算的 VIP、EA 和可用的实验值。 Ag n 的计算 VIP 和 EA + 1 个簇与其实测数据相符。这种一致性再次证实了当前理论方法的可靠性。此外,我们注意到 AgV 二聚体具有最大的 VIP 和最小的 EA。这意味着 AgV 很难丢失或需要电子。二十面体 Ag12V 簇拥有最大的 EA,很容易再得到一个电子。为以后光电子能谱实验提供参考资料,Ag n 基态和两个低位结构的理论光电子能谱(PES) V (n =1-12) 簇的模拟方法是将第一个 VIP 添加到每个相对于 HOMO 的占据轨道能量,并用洛伦兹展开方案和 0.1 eV 的展宽因子拟合它们,如图 4 所示。 能级分布这些簇中的一个在 5.5 到 12 eV 的范围内。实验者可以利用PES光谱来区分这些簇。
<图片>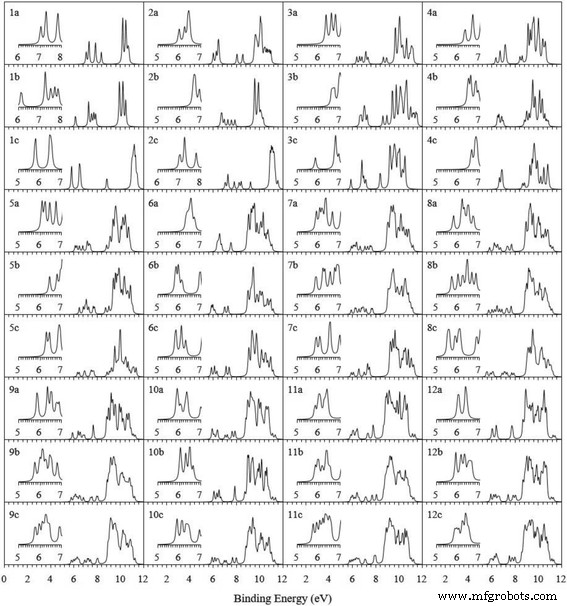
Ag n 基态和两种低位异构体的模拟 PES V (n =1-12) 个簇
为了检验 V 原子对银团簇稳定性的影响,原子平均结合能 (E b) 最稳定的Ag n + 1 和 Ag n V簇可以估计如下:
$$ {E}_b\left({\mathrm{Ag}}_{n+1}\right)=\left[\left(n+1\right)E\left(\mathrm{Ag}\right) -E\left({\mathrm{Ag}}_{n+1}\right)\right]/\left(n+1\right), $$ (3) $$ {E}_{\mathrm{ b}}\left({\mathrm{Ag}}_n\mathrm{V}\right)=\left[ nE\left(\mathrm{Ag}\right)+E\left(\mathrm{V}\right) )-E\left({\mathrm{Ag}}_n\mathrm{V}\right)\right]/\left(n+1\right), $$ (4)其中 E (Ag), E (Ag n + 1), E (V) 和 E (Ag n V) 分别是 Ag 原子、银团簇、V 原子和 AgnV 团簇的能量。最稳定的 Ag n 每个原子的计算结合能 + 1 和 Ag n V 簇绘制在图 5 中。从该图中可以清楚地看出 E b Ag n V簇是簇大小的单调递增函数,大于Ag n + 1 个用于 n 的集群 ≥ 2. 特别是E b 对于平面结构,掺杂簇的数量迅速增加,而对于 3D 结构,则逐渐增加。这意味着原子之间的键合力在生长过程中变得越来越强。 Ag n 中一个 V 原子取代一个 Ag 原子 + 1(n ≥ 2) 集群可以明显提高宿主集群的稳定性。另一方面,双原子团簇的键能应与键长密切相关。 E AgV 二聚体的 b 小于 Ag2。这种异常变化可能是由于AgV的键距(2.61 Å)比Ag2的键距(2.58 Å)长。
<图片>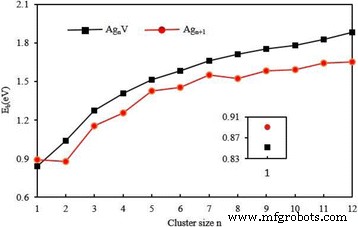
最低能量Ag n的平均结合能 + 1 和 Ag n V (n =1-12) 个簇
团簇的热稳定性可以通过解离能 (DE) 来检查,这对于不同的解离通道是不同的。最基本的解离通道是将一个较大的簇分裂成两个较小的簇。相对于其他解离通道,相应的 DE 较小。因此,研究了最稳定的 Ag n 的后续解离通道 V (n =1-12) 簇。
$$ {\mathrm{Ag}}_n\mathrm{V}\to {\mathrm{Ag}}_m+{\mathrm{Ag}}_{n-m}\mathrm{V} $$ (5)其中 m 不超过 n .上述解离通道的DE定义如下:
$$ {\mathrm{DE}}_m\left({\mathrm{Ag}}_n\mathrm{V}\right)=E\left({\mathrm{Ag}}_m\right)+E\left( {\mathrm{Ag}}_{nm}\mathrm{V}\right)-E\left({\mathrm{Ag}}_n\mathrm{V}\right) $$ (6)其中 E 代表相应簇或原子的能量。 Ag n 的 DE 表 4 中列出了不同解离通道的 V 簇。小 DE 表示相应的解离通道容易发生。也就是说,最小DE对应的解离通道最有可能出现。从表 4 可以看出,Ag n 最优选的解离通道 V簇是Ag n V =Ag + Ag n - 1V for n =1 和 4-12 和 Ag n V =Ag2 + Ag n - 2V for n =2和3。Ag12V簇的最小DE(2.54eV)在所有掺杂簇中最大,这意味着二十面体簇比其他簇更稳定。此外,我们发现 3D 中性 Ag n 的最小 DE 的变化趋势 V (n =5–12) 簇与阳离子 Ag n 的丰度相同 V + 集群 [64, 65]。然而,平面 Ag n 之间没有这种关系 V 和 Ag n V + 对于 n =2–4。
能隙 (E g) 最高占据分子轨道 (HOMO) 和最低未占据分子轨道 (LUMO) 之间的差值始终被认为是表征小金属簇化学活性的重要量。大的能隙与高化学稳定性有关。对于基态 Ag n + 1 和 Ag n V 簇,图 6 显示了作为簇大小函数的能隙。在纯银团簇的能隙中观察到奇偶交替。这种交替可以用电子配对效应来解释,即占据相同 HOMO 的两个电子的电子屏蔽效应远小于占据不同轨道的两个电子的屏蔽效应。一个 Ag 原子 ([Kr]4f 14 4d 10 5s 1 ) 在 Ag n + 1 个簇被 V ([Ar]3d 3 4s 2 ) 原子。对于奇数 n , Ag n 的闭壳 + 1 个簇被 Ag n 的开壳取代 V簇。当然,E g 银 n 具有奇数 n 的 V 簇 小于 Ag n + 1 个集群。这种下降是非常明显的。对于偶数 n , 都 Ag n + 1 和 Ag n V 群集具有不受限制的外壳。 E g 应该取决于它们的结构。在这种情况下,我们注意到 E g 银 n V (n =2 and 4) 平面结构的团簇小于Ag n + 1 个集群和 E g 银 n V (n =6, 8, 10, 12) 3D结构的簇比Agn稍大 + 1 个集群。一般来说,Ag n 中一个 V 原子取代一个 Ag 原子 + 1 个具有偶数 n 的簇 对宿主簇的能隙影响不大。
<图片>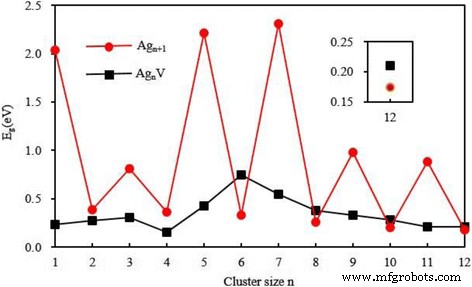
基态 Ag n 的 HOMO-LUMO 能隙 + 1 和 Ag n V (n =1-12) 簇
磁性属性
团簇的磁性常用于制备纳米电子器件和高密度磁存储材料。簇的总磁矩由电子的自旋磁矩和轨道磁矩组成。电子的自旋磁矩远大于轨道磁矩,因此簇的磁矩由自旋磁矩支配。最低能量 Ag n 的总磁矩 V 个簇 (n =1-12) 簇已被计算并显示在图 7 中,其中我们还绘制了宿主簇的总磁矩。最稳定的 Ag n 的磁矩 + 1 个簇被奇数 n 完全淬灭 并且对于偶数 n 是 1 μB .小银 n V簇具有很大的磁矩。随着团簇尺寸的增加,Agn的磁矩 V簇在波浪中减少。当 n =12,Ag12V 与 Ag13 团簇具有相同的磁矩。这意味着 V 原子的掺杂只能增强小银团簇的磁性。为了解释磁性,图 8 显示了基态 Ag n 的自旋态密度 (SDOS) V簇。从图中可以看出 Ag n V簇有一些磁畴随着簇大小的增加而减小。所有最低能量结构在− 5 eV 和− 2.5 eV 之间都有一个强能带,主要由价s 组成 和 d Ag 和 V 原子的轨道。 HOMO 附近的能级,E − E HOMO =− 1~0 eV,在确定 Ag n 的磁行为中起关键作用 V簇。
<图片>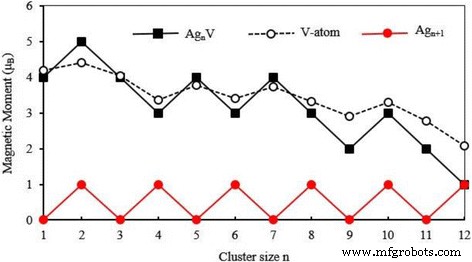
基态总磁矩 Ag n + 1 和 Ag n V (n =1-12) V 原子上的簇和局部磁矩
<图片>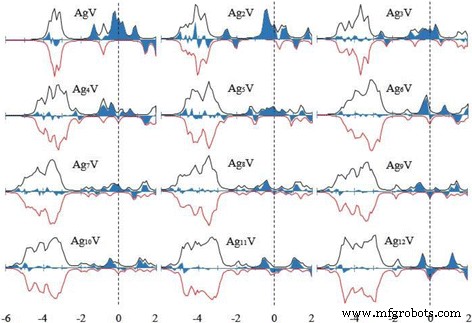
基态 Ag n 的 SDOS V (n =1-12) 簇。向上旋转为正,向下旋转为负。使用加宽因子 δ =0.1 eV。向上旋转减去向下旋转是蓝色部分。虚线表示HOMO能级的位置
为了进一步探索磁性,我们对最稳定的Ag n进行了自然键轨道分析 V簇[66]。 V原子上的局部磁矩为4.18 μ B 代表 AgV,4.41 μ B 代表 Ag2V,4.03 μ B 代表 Ag3V,3.36 μ B 代表 Ag4V,3.78μ B 为 Ag5V,3.40 μ B 代表 Ag6V,3.73 μ B 代表 Ag7V,3.33 μ B 代表 Ag8V,2.91 μ B 代表 Ag9V,3.29 μ B 为 Ag10V,2.77 μ B 为 Ag11V,和 2.08 μ B 为 Ag12V,如图 7 所示。总体而言,V 原子的磁矩随着团簇尺寸的增加而逐渐减小。 Ag原子提供的磁矩非常小。此外,除了 Ag2V、Ag5V 和 Ag7V 簇外,其他掺杂簇中 Ag 原子的总磁矩相对于 V 原子的磁矩表现出反铁磁排列。换句话说,所有 Ag n 的总磁矩 V簇主要来源于顺磁性V原子,如图7所示。
4s上的局部磁矩和电荷 , 3d , 4p , 和 4d 最低能量 Ag n 中的 V 原子壳 V簇列于表5中。从该表中可以看出部分占用的3d 壳在决定V原子的磁性中起关键作用,其磁矩为2.01~3.82 μ B. 4s 和 4p 壳对于自由 V 原子是非磁性的,会产生一点磁矩。 4d 外壳几乎没有磁性。 3d 上的电荷 和 4p 炮弹增加 0.77–1.97 和 0.03–2.41 e 分别。尤其是 4p 上的电荷 轨道随着簇大小的增加而增加。在 4d 上发现很少的电荷 V 原子在 Ag n 中的轨道 V (n =4–12) 簇。尽管如此,4s 上的电荷 shell 减少了 1.02–1.54 e .电荷转移表明 Ag n 中的 V 原子 V簇在s之间有杂交 , p , 和 d 贝壳。众所周知,孤立的 V 原子有五个价电子。同时,Ag n 中 V 原子的电荷 V簇可以从表5中得到。从电荷守恒原理,0.10-0.21 e 平面 Ag n 从 V 原子转移到 Ag 原子 V (n =1–4) 集群,而 0.35–2.92 e 3D Ag n 从 Ag 原子到 V 原子 V (n =5–12) 个簇,如图 9 所示。如果 M 和 C 表示 Ag n 中 V 原子的磁矩和价电子 V簇,磁矩的变化(ΔM =M − 3) 和电荷转移 (ΔC =5 − C ) 具有相同的变化趋势,如图 10 所示。从图 10 可以得出结论,电荷转移应该是 Ag n 中 V 原子磁矩改变的原因 V簇。
<图片>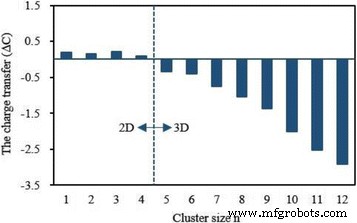
最稳定的Agn中V原子的电荷转移 V (n =1–12) 簇。自由V原子作为参考点
<图片>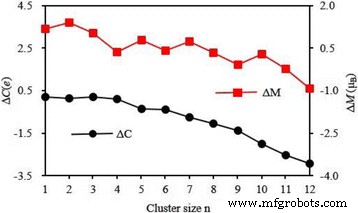
最稳定的Ag n中V原子的电荷转移(ΔC)和磁矩(ΔM)变化 V (n =1–12) 簇
结论
Ag n 的结构、电子和磁性特性 V (n =1-12) 簇已经在 DFT 和 CALYPSO 结构搜索方法的基础上进行了研究。结果表明V原子在最低能量Agn V簇往往占据协调数最高的位置。 Ag n 中 Ag 原子的取代 + 1 (n ≥ 5) 一个 V 原子簇改变了宿主簇的几何形状。 Ag n的红外光谱、拉曼光谱和PES V (n =1-12) 簇有望在未来识别基态。除了 AgV,其他 Ag n 的稳定性 V簇高于Ag n + 1 个集群。相对容易解离的通道是 Ag n 对于n,V =Ag + Agn − 1V =1 和 4-12 和 Ag n V =Ag2 + Ag n - 2V for n =2和3.Agn的化学活性 具有奇数 n 的 V 簇 高于Ag n + 1 个集群。 Ag n 的磁矩 V簇主要来源于掺杂的V原子,从5到1μ逐渐减少 B 随着集群规模的增加。磁矩的变化可能是由于V原子与Ag原子之间的电荷转移所致。
缩写
- 3D:
-
三维
- DE:
-
解离能
- DFT:
-
密度泛函理论
- EA:
-
电子亲和性
- HOMO:
-
最高占据分子轨道
- LUMO:
-
最低未占分子轨道
- PSO:
-
Particle swarm optimization
- VIP:
-
Vertical ionization potential
纳米材料