

过渡金属掺杂高岭石纳米粘土的结构和电子特性
摘要
在这项工作中,通过密度泛函理论 (DFT) 计算研究了一系列过渡金属(Cr、Mn、Fe 和 Co)掺杂的高岭石纳米粘土。分析了金属掺杂对高岭石几何结构和电子结构的影响。研究了过渡金属 (TM) 掺杂高岭石结构的铁磁性 (FM)、反铁磁性 (AFM) 和非磁性 (NM) 状态。通过色散校正密度泛函理论 (DFT-D2) 计算晶体体积、晶格参数、键长、电荷和自旋。结果表明,Cr 3+ 和 Fe 3+ 掺杂剂在 AFM 状态下表现更稳定,而 Mn 3+ 首选 AFM 和 FM 状态,以及 Co 3+ 掺杂剂首选 NM 状态。此外,过渡金属掺杂可以引起晶格体积膨胀和带隙中的一些掺杂状态。
背景
高岭土类纳米粘土矿物由于水热蚀变和/或风化过程而具有独特的物理性质,因为它们具有层状结构、粒度小,最重要的是水合表面含有大量羟基。它引起了材料化学、环境化学和矿物物理学研究人员的关注[1,2,3,4,5,6,7,8,9,10,11]。高岭石是地球上最丰富的纳米粘土矿物之一,已广泛用于塑料、催化和水泥工业。高岭石作为新型载体材料的进一步功能化越来越受到各个领域的关注。高岭石可以简单地作为载体材料与其他纳米粒子混合形成用于太阳能的相变材料 [4, 5] 或涂有掺杂氧化物以形成用于导电领域的导电粉末 [9, 12]。高岭石与功能性纳米颗粒的杂化被发现通过协同效应提高了 Pd-ZnO 的光催化活性和 CdS 的发光性能 [6, 7]。通过在表面锚定一些官能团[13, 14]或通过酸活化预处理进一步改善高岭石的表面性质[2]。
高岭土族矿物的结构和能量学已通过实验 [15,16,17] 和理论 [18,19,20,21,22] 进行了广泛研究。高岭石表面重金属吸附理论研究对Cd、Cu、Hg和Ni(II)的吸附进行了研究[23],其中高岭土对离子的吸附能力依次为Ni>Cu>Cd>汞(Ⅱ)。研究了Pb(II)[24, 25]和铀酰[26]在高岭石(001)表面的吸附和扩散[24,25,26],其在水体系中的吸附行为也有报道[27, 28]。研究了 Mg、Ca 和 Fe 掺杂对高岭石表面的影响,以及随后 H2O 对夹层的吸附和渗透[29]。发现 H2O 在掺杂高岭石 (001) 上的吸附能小于未掺杂表面。标准密度泛函理论 (DFT) 泛函和混合泛函 [30] 研究了具有和不具有固有缺陷的高岭石的电子结构。然而,直到最近,高岭石的脱羟基、脱铝和二氧化硅缩合过程中的结构演变才通过 DFT 计算进行建模 [1, 31, 32]。高岭土族材料中Al的去除极大地改变了这些层状材料的几何形状和电子性质,提高了它们的支撑效果[1, 2]。
金属掺杂作为一种众所周知的改变化合物结构和性质的方法,已经在理论上研究了 Al2O3 [33]、TiO2 [34]、MOF [35] 和其他固体 [36]。探索过渡金属 (TM) 掺杂后高岭石纳米粘土的结构和性质的变化对于这种层状粘土材料将是有趣的。本工作通过DFT计算研究了一系列Cr、Mn、Fe和Co掺杂的高岭石纳米粘土,重点研究了金属掺杂对高岭石纳米粘土几何结构和电子结构的影响。研究了这些过渡金属掺杂的高岭石结构的可能的铁磁性 (FM)、反铁磁性 (AFM) 和非磁性 (NM) 状态。采用色散校正密度泛函理论(DFT-D2)对晶格参数、键长、电荷和自旋进行优化计算。
方法
所有计算均使用程序 CASTEP(剑桥顺序总能量包)代码 [37],基于第一原理 DFT。 Perdew、Burke 和 Ernzerhof (PBE) 使用具有交换相关电位的广义梯度近似 (GGA) 进行计算 [38]。包括 Grimme 的 DFT-D2 色散校正以解释范德华色散相互作用 [39]。使用超软赝势平面波形式化 [40] 应用 500 eV 的能量截止。 Monkhorst–Pack [41] 网格具有 2 × 2 × 3 k 点网格用于几何松弛和电子结构计算。通过密度混合方案[42]有效地获得了基态的自洽总能量。对于几何优化,自洽场 (SCF) 容差的收敛阈值设置为 1.0 × 10 −6 eV/atom,原子上的所有力都收敛到小于 0.03 eV/Å,总应力张量降低到 0.05 GPa 量级,最大离子位移在 0.001 Å 以内。价态元素为O(2s 2 2p 4 ), Al(3s 2 3p 1 ), Cr(3s 2 3p 6 3d 5 4s 1 ), Mn(3d 5 4s 2 ), Fe(3d 6 4s 2 ), 和 Co(3d 7 4s 2 )。 Mn、Fe 和 Co 使用 uspcc 赝势,其余元素使用 usp 赝势。在使用 Broyden-Fletcher-Goldfarb-Shanno (BFGS) 最小化算法进行几何优化期间,电池参数和原子配位完全放宽。通过对TM离子施加不同的初始磁矩来消除晶体对称性,从而使电子基态采用较低的对称性。
结果与讨论
我们之前的工作采用了最初的高岭石结构 [1]。图1显示了高岭石(4个高岭石单元)的松弛2 × 2 × 1晶体结构。高岭石层状结构 Al2Si2O5(OH)4 由八面体 Al-O 片和四面体 Si-O 片组成,由顶部 O 原子 (Oa) 连接。 Si-O四面体由一个中心Si原子和四个周围的O原子构成,其中一个是Oa原子,另外三个是基础O原子(Ob)。 Al-O 八面体由一个中心 Al 和六个周围的 O 构成,其中两个是 Oa 原子,另外四个是与其他 Al-O 八面体共享的 O 原子(在 OH 基团中)。此外,这些OH基团可分为两种:层结构表面的层间OH(OHinter)和Al片和Si片之间层结构内部的内部OH(OHinner)。因此,有两种 Si-O 键,Si-Oa 和 Si-Ob(黑点线),以及三种 Al-O 键,Al-Ointer(红点线),Al-Oinner(绿点线) )、高岭石块体结构中的Al-O(黑点线)。
<图片>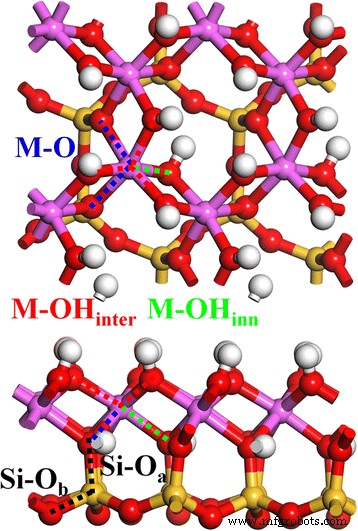
顶部(向上 ) 和侧面 (向下 ) 高岭石的意见。 Si-Oa(黑色 ), Si–Ob (黑色 ), M–OHinter (红色 ), M–OHinner (绿色 ), 和 M–O (蓝色 ) 键用点线表示
由于层间的相互作用,弥散能在粘土矿物的结构稳定中始终起着重要作用[21, 43]。在几种混合泛函中,PBE-D2 [21]、B3LYP [22]、B3LYP-D [18] 和 RPBE-D2 [18, 21],用于获得高岭石的实验晶格结构 [44, 45] ],发现 PBE-D2 功能既准确又耗时。正如之前简要报道的那样,与实验结果相比,通过色散校正克服了对键长的 PBE 函数的高估 [1]。为了区分 TM 掺杂对高岭石结构的影响,我们首先重新审视晶格结构以及中心阳离子(Si 和 Al)与氧原子 Oa、Ob 和 OHinn 之间的优化键距。
如表 1 所示,对于高岭石,使用色散校正的 PBE-D2 泛函优化的计算晶胞体积接近实验值,与 PBE 泛函(~3.4%)相比,相对误差(~0.4%)显着降低.对于晶格向量 a 和 b,使用 PBE-D2(~0.4%)的相对误差远低于 PBE(~1.1%)。并且,在 PBE-D2 的色散校正下,高岭石的层距(矢量 c)减少了 0.17 Å(~2%)。值得注意的是,色散校正后的晶格角与实验结果非常接近,尤其是对于 α。至于高岭石中的键长分布,虽然与实验结果相比,PBE-D2 对 Si-Oa、Al-OHinner 和 Al-O 键的改善不大,但对 Al-O 表面的 Al-OHinter 键有很大的改善(这对表面化学很重要)和 Si-O 表面的 Si-Ob 键的轻微改善。值得注意的是,对于 Al-OHinter 键,来自 PBE-D2 的色散校正似乎准确地描述了 Al-O 表面最外层的键合环境,这受到另一个高岭石层的 Si-O 表面的色散力的强烈影响就在上面。这里要提到的另一点是实际上有两个分裂的 Al-O 键(图 1,蓝色点线),键长显着不同,约为 1.95 和 2.00 Å [45],这显示了 Al-O 的晶格畸变八面体起源于 Si-O 片和 Al-O 片之间的晶格失配。与实验结果相比,高岭石结构计算的一个主要错误是,这些 Al-O 键被 PBE 和 PBE-D2 高估,具有相似的平均键长(表 1)。 PBE-D2 提供了大约 1.96 和 2.04 Å 的两个 Al-O 键,第二个键高估了 0.04 Å(图 2,蓝色点线)。
<图片>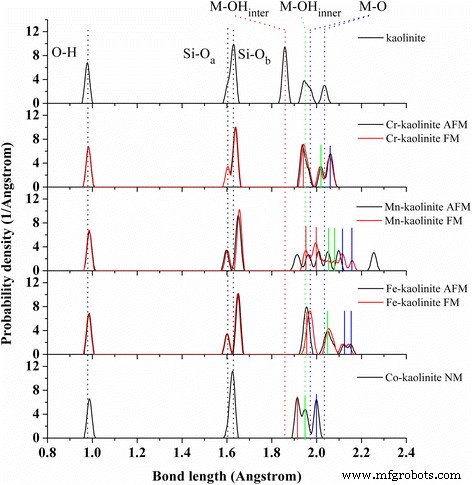
Cr-、Mn-、Fe- 和 Co-高岭石的键分布。每个 TM-高岭石都给出了多磁态。平均不同类型的 O–H(黑色 ), Si-Oa (黑色 ), Si–Ob (黑色 ), M–OHinter (红色 ), M–OHinner (绿色 ), 和 M–O (蓝色 ) 高岭石中的键用点线表示 . M-OHinter(红色 ), M–OHinner (绿色 ), 和 M–O (蓝色 ) Cr-高岭石 (AFM ), Mn-高岭石 (FM ), Fe-高岭石 (AFM ), 和 Co-高岭石 (NM ) 用实线表示
过渡金属(Cr、Mn、Fe 和 Co)掺杂的高岭石是通过用 Cr、Mn、Fe 或 Co 原子替换 Al 原子来构建的。只有Al 3+ 的等价置换 离子与 TM 3+ 离子被认为是因为 TM 离子的非等价取代化学状态不是 +3 会导致额外的空位或杂质以达到电荷平衡。从结构的角度来看,TM-高岭石的 PBE 和 PBE-D2 官能团给出了与高岭石观察到的相似的结构差异。考虑到 PBE-D2 泛函对高岭石两个基面的晶格矢量和键长描述得更好,下面讨论 TM-高岭石,主要取决于 PBE-D2 泛函获得的结果。表 1 总结了 TM 掺杂高岭石的晶格参数、键长、电荷和自旋及其磁态。 Cr-高岭石、Mn-高岭石和 Fe-的 AFM 和 FM 状态之间的能量差异(每个 TM 原子)高岭石分别为 0.022、-0.006 和 0.094 eV。由于Co-高岭石结构仅在非磁性状态下稳定,因此仅显示Co-高岭石的NM结构。
与高岭石相比,TM-高岭石的晶胞体积扩大,趋势为Mn-高岭石> Fe-高岭石>> Cr-高岭石>> 高岭石> Co-高岭石。细胞膨胀主要是由于与 Al-O 键相比更长的 M-O 键引起的,导致晶格矢量 a 和 b 的主要膨胀。同时,Si-O片上的Si-Ob键同时被拉长,α和β的晶格角相应地扭曲。与 AFM 状态相比,具有 FM 状态的 Mn-高岭石的晶胞体积增加了 1.4%,而相比之下,磁性排序对 Cr-高岭石和 Fe-高岭石的晶胞体积几乎没有影响。 Cr、Mn、Fe和Co的磁矩与TM掺杂Al2O3的磁矩接近[33],而Mulliken电荷略高,表明反应性更强。
图 2 分析了 TM-高岭石的键长分布,TM-高岭石中不同类型的 Si-O 和 M-O 键用实线表示每种掺杂元素。总体而言,TM掺杂后M-O和Si-Ob的键长增加,同时M-OHinter(红色),M-的分裂M-O键的键分布重组OHinner(绿色)和 M-O(蓝色)键。值得注意的是,在 Cr 和 Co 掺杂后,分裂的 Al-O 键(蓝点线)消失了。此外,键长分布高度依赖于Mn原子的磁序,而对Cr和Fe原子的影响很小。
Cr 3+ 的 PDOS 结果 (d3), Mn 3+ (d4), Fe 3+ (d5) 和 Co 3+ (d6) 和相应的电荷密度分布如图 6 和图 6 所示。 3 和 4。根据 Jahn-Teller 定理,任何退化的电子系统都会自发地扭曲,以消除受到周围键合环境 [47] 影响的退化 [46]。对于 TM 3+ 掺杂大量羟基的高岭石八面体Al位点,TM 3+ 的五个d-shell轨道 在 Oh 对称性下将分裂成三重态 t2g 状态和双重态 eg 状态。三重态的电子位于配体之间的中间区域,并进一步与最近的 O 态杂交。处于双重态的那些直接指向配体,因此比 t2g 电子具有更高的能量。通常,电子在 eg 轨道中的存在往往会使八面体键不稳定,并且通过延长与填充轨道相对的键并缩短与空轨道相对的键来消除简并性。 TM 3+ 的d-d跃迁 (哦)物种总是从占据的 t2g 轨道(dxy、dyz 和 dzx)到未占据的例如轨道(dx2-y2 或 dz2,取决于它们的占有率)。 Cr 3+ 的 eg 轨道和 t2g 轨道之间的轨道分裂 (d 3 ), Mn 3+ (d 4 ), Fe 3+ (d 5 ), 和 Co 3+ (d 6 ) 在 TM-高岭石中与在 Al2O3 和 TiO2 中的相似 [33, 48, 49],但 3d 轨道之间的分裂能略大于它们自己的氧化物(图 3),可能是由于与周围的杂化羟基。
<图片>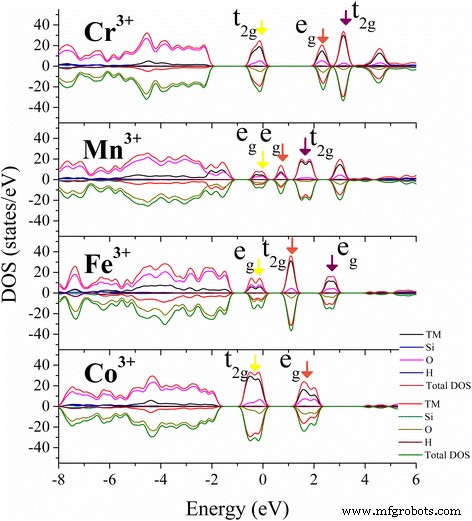
给出了 TM 掺杂高岭石的最稳定态的总态密度 (DOS) 和原子投影态密度 (PDOS)。最高占据的 3d 轨道(黄色 ) 和第一个 (brown ) 和第二个 (紫色 ) 费米能级周围最低的未占据 3d 轨道由 彩色箭头 指向
<图片>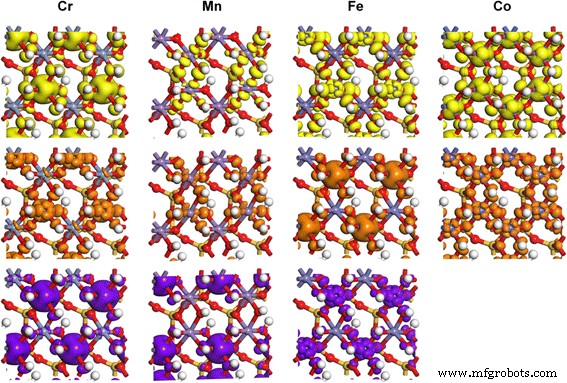
TM-高岭石中TM 3d轨道的部分电荷密度,对应于箭头所指的状态 在 PDOS 结果中。等值面水平为 0.02 e/Å 3
Mn-高岭石的FM和AFM状态之间的分裂能差异很小,除了自旋方向不同外,态密度分布相似。因此,为简单起见,仅显示了 AFM 状态的结果。对于高自旋 Mn 3+ (d 4 ) 离子在具有 AFM 状态的 Mn-高岭石中,只有两个 eg 轨道之一被占据在价带最大值 (VBM)(图 3,黄色箭头)。占据能量较低的 dz2 轨道对沿 z 的两个配体的键合电子产生强烈排斥 轴并在该方向拉长 M-O 键合。这种效应就是著名的 Jahn-Teller 效应。导带最小值 (CBM) 底部的状态由最低未占据的 dz2 轨道(棕色箭头)和 Mn 3+ 的较高 dx2y2 轨道(紫色箭头)组成 (d 4 )。对于 Cr 3+ (d 3 ), Fe 3+ (d 5 ), 和 Co 3+ (d 6 ) 掺杂的情况下,其中 t2g 和 eg 轨道均匀占据,Jahn-Teller 畸变效应的影响很小,仅导致 TM-高岭石中 M-O 键的轻微偏差(图 2)。通过TM掺杂对结构和电子性质的这种改变可能会改善高岭土在催化[50, 51]、CO捕获[52, 53]、载药[54]和储能[55,56,57]等领域的应用]。并且,它也可以应用于其他矿物,如蒙脱石[50, 58]、珍珠岩[55]和滑石[59]以改变它们的电子性质。
结论
通过DFT计算研究了过渡金属(Cr、Mn、Fe和Co)掺杂对高岭石纳米粘土几何结构和电子结构的影响。计算和研究了晶体体积、晶格参数、键长、电荷和自旋以及可能的磁态。 Cr 3+ 和 Fe 3+ 掺杂剂在 AFM 状态下表现更稳定,Mn 3+ 更喜欢 FM 状态,并且 Co 3+ 掺杂剂更喜欢 NM 状态。过渡金属掺杂引起晶格体积膨胀和 M-O 键分布的一些重组。同时,TM掺杂剂在高岭石的带隙中引入了一些具有较大分裂能的3d态。
缩写
- 原子力显微镜:
-
反铁磁性
- BFGS:
-
Broyden-Fletcher-Goldfarb-Shanno
- CASTEP:
-
剑桥顺序总能量包
- CBM:
-
导带最小值
- DFT:
-
密度泛函理论
- DFT-D2:
-
色散修正密度泛函理论
- 调频:
-
铁磁性
- GGA:
-
广义梯度逼近
- NM:
-
无磁性
- PBE:
-
Perdew、Burke 和 Ernzerhof
- SCF:
-
自洽字段
- TM:
-
过渡金属
- VBM:
-
价带最大值
纳米材料