

来自第一性原理研究的 SiAs 和 SiAs2 单层中的应变可调带隙和高载流子迁移率
摘要
寻找新的稳定的独立原子级薄二维 (2D) 材料对当代材料科学的基础和实践方面具有重要意义。最近,已经实现了层状 SiAs 单晶的合成,这表明它们的少数层结构可以被机械剥离。执行第一性原理密度泛函理论计算,我们提出了两种动态和热力学稳定的半导体 SiAs 和 SiAs2 单层。能带结构计算表明,它们都表现出间接带隙,并且通过施加应变发现了间接到直接带甚至到金属过渡。此外,我们发现 SiAs 和 SiAs2 单层比 MoS2 具有更高的载流子迁移率,并显示出像黑色磷烯一样的各向异性传输,使其在光电子学中有潜在的应用。我们的工作为纳米级光学器件的新功能开辟了一条新途径。
背景
原子级薄的二维 (2D) 晶体已成为当代材料科学发展最快的领域之一。多功能的电子特性、出色的电子迁移率以及在纳米电子学和光电子学中的有前景的应用,正在推动大部分凝聚态物理学家寻找新的二维材料。继石墨烯 [1-4] 之后,还合成了大量其他二维材料,例如硅烯 [5-7]、氮化硼纳米片 [8、9]、过渡金属二硫属元素化物 (TMD) [10、11]、黑磷 [12, 13]、硼烯 [14-16]、砷烯 [17, 18]、碲烯 [19] 及其等电子化合物 [20-23]。二维材料的列表正在迅速扩大,现在已知的此类材料有数千种,涵盖了电子和其他特性的全部范围。并且它们的新特性不同于甚至优于它们的本体,在理论上得到了预测和实验证实。
尽管在寻找各种二维材料(包括一些已经具有带隙或其他理想特性的材料)方面投入了大量大量精力,但尚未达成共识。石墨烯具有惊人的载流子迁移率、高机械稳定性和无质量狄拉克电子,迄今为止备受关注,但缺乏固有带隙阻碍了其在现代电子器件工业中的应用。尽管已经做出了很大的努力,但还没有达到在没有副作用的情况下打开相当大的带隙 [24, 25]。在光电器件中具有高性能的 TMD 确实具有固有带隙,但载流子迁移率较差 [26-28]。具有应变敏感的可调带隙和各向异性高载流子迁移率的黑磷和蓝磷在空气中不能保持稳定 [13, 29]。最近,已经实现了层状SiAs和SiAs2单晶的合成[30-32],这表明通过机械剥离可以获得很少的层状结构。
在目前的工作中,基于第一性原理密度泛函理论计算 (DFT),我们提出了两种动态和热力学稳定的半导体单层 SiAs 和 SiAs2。它们都具有间接带隙(分别为 2.39 eV 和 2.13 eV)。沿两个面内方向施加各向同性应变实际上将 SiAs (SiAs2) 单层转化为直接间隙 1.75 eV (1.60 eV) 材料。此外,我们发现 SiAs 和 SiAs2 单层比 MoS2 具有更高的载流子迁移率,并显示出像黑色磷烯一样的各向异性传输,使其在光电子学中有潜在的应用。我们的工作为纳米级光学器件的新功能开辟了一条新途径。
计算方法
DFT 计算是使用 Vienna ab initio 模拟包 (VASP) 代码 [33] 进行的。我们在广义梯度近似(GGA)下使用了 Perdew-Burke-Ernzerhof (PBE) [34] 交换相关函数。投影仪增强波 (PAW) 方法 [35] 用于描述电子 - 离子相互作用。施加垂直于片材(沿 c 轴)的 20 Å 真空以避免层间相互作用。 500 eV 的动能截止值用于平面波基组。布里渊区采样是使用 15 × 5 × 1 Monkhorst-Pack [36] 网格进行的,用于 2D 片材。用于电子自洽弛豫和离子弛豫的收敛标准设置为 10 -4 能量和力分别为 0.01 eV/Å。声子计算是通过 PHONOPY 代码使用超胞法进行的 [37, 38],并且在 VASP 中实施的密度泛函微扰理论 (DFPT) 中计算了超胞的真实空间力常数。此外,更严格的能量(10 −8 eV/atom) 和力收敛准则 (10 −4 eV/Å) 用于振动谱计算。在分子动力学(MD)计算中,采用(3×3×1)个超胞,在摩尔-体积-温度(NVT)系综中,温度保持在 300 K 6 ps,时间步长为 2 fs。使用CASTEP代码[39-41]在PBE理论水平计算拉曼光谱。
结果和讨论
松弛的独立式 2D SiAs 和 SiAs2 的几何结构和电子密度分布分别显示在图 1a、b 中,它们的体结构显示在附加文件 1:补充材料的图 S1 中。如附加文件 1 所示:图 S1a 和 b,块体 SiAs(SiAs2) 具有 C2/m(Pbam) 对称性,由堆叠的 Si-As 层组成,由范德华力弱结合,距离为 3.06 Å (1.66 Å) )。单层SiAs的晶胞为菱形,优化后的晶体参数为a 1 =3.69Å 和 b 1 =10.83Å 与 φ =99.81°。 SiAs包含6个Si原子和6个As原子。每个 Si 原子有四个最近的相邻原子(3 个 As 和 1 个 Si),而每个 As 原子仅与相邻的 Si 原子形成三个共价键。它存在两种键,即Si-Si键和Si-As键。 Si-Si键长约为2.35 Å,Si-As键长在2.39 Å和2.43 Å之间,屈曲高度为d 1 =4.86 埃。在单层 SiAs 的侧视图中,形成了双层和单层交替膨胀的眼镜绳状结构。另一种硅和砷化合物的单层结构是SiAs2。其主晶胞包含4个Si原子和8个As原子,呈矩形结构,优化后的晶体参数为a 2 =3.68Å 和 b 2 =10.57 埃。每个 As 原子有三个最近的相邻 Si 原子或与相邻的 Si 原子形成一个共价键和两个共价键,而每个 Si 原子只有四个最近的相邻 As 原子。与前者不同,SiAs2 拥有较弱的 As-As 键 (2.50 Å) 而不是 Si-Si 键。其Si-As键范围为2.41 Å至2.45 Å,屈曲高度为d 2 =5.09 埃。从电子密度分布来看,As原子由于其电负性大而从Si原子中吸引电子,具有较大的电子密度。为了协助未来的实验表征,我们进一步计算和检查了体和单层 SiAs 和 SiAs2 的拉曼光谱。在附加文件 1:补充材料的图 S2 中可以看到单层和全晶体之间的明显变化,其起源已被确定为层范德瓦尔斯相互作用的影响 [42]。
<图片>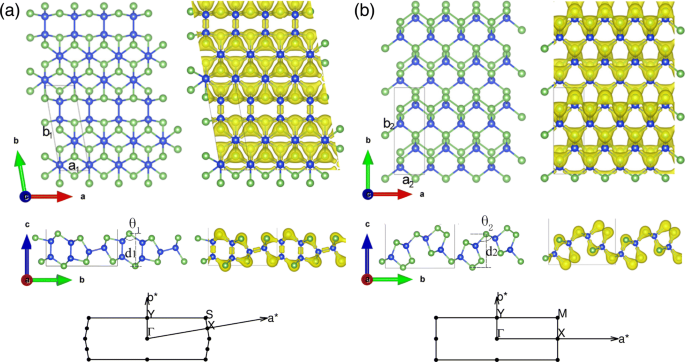
单层 SiAs 和 SiAs2 的几何结构和电子密度分布。 (在线颜色)单层的俯视图和侧视图 a SiAs和b SiAs2 几何结构和电子密度分布以及相关的布里渊区。蓝色和绿色的球分别表示 Si 和 As 原子
为了了解 SiAs (SiAs2) 的稳定性,我们首先计算了内聚能,定义为 E coh =(nE Si + mE As-E 单声道)/(n + 米 ),其中 E 硅,E 作为,和 E Mono分别是单个Si原子、单个As原子和单层SiAs(SiAs2)的一个分子式单元的总能量,n(m)是分子式单元中As(Si)原子的数量。我们的计算表明,SiAs 单层的内聚能为 5.13 eV/atom,比 SiAs2 单层的 4.98 eV/atom 稍大。相比之下,在相同的理论水平下,砷烯和硅烯的内聚能分别为 2.99 和 3.71 eV/atom [18, 43]。 SiAs和SiAs2的高内聚能表明两者结合牢固且稳定性高。
为了进一步确认单层 SiAs 和 SiAs2 的结构稳定性,我们还进行了振动声子光谱计算。如图 2a 所示,除了 Γ 附近的横向声学模式外,正频率占大多数模式 点,这是由于声子的软化,并已在其他类似系统中报道 [44, 45],表明结构都是动态稳定的。然后,我们在室温 (T =300K ),如图 2b 所示。轻微的能量波动和保存完好的结构表明它们在室温下是热稳定的。我们的结果表明单层SiAs和SiAs2可以在室温下实现实验。
<图片>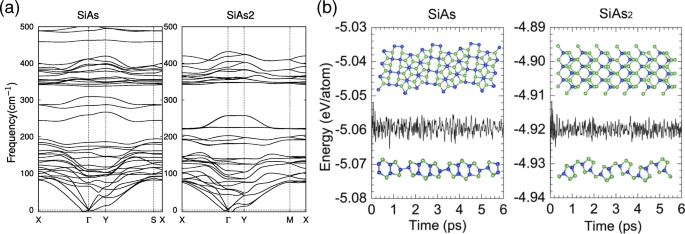
单层 SiAs 和 SiAs2 的声子色散曲线和 MD 模拟。 一 单层 SiAs 和 SiAs2 的声子色散曲线。 b SiAs 和 SiAs2 室温 MD 模拟过程中总能量和时间的关系。还提供了 6 ps 结束时单层结构的精选快照
随着单层 SiAs 和 SiAs2 的优化结构,现在我们关注它们的电子特性。 SiAs和SiAs2单层的计算轨道分解能带结构如图3所示。我们的计算清楚地表明SiAs和SiAs2单层都是具有宽带隙的间接半导体。对于单层 SiAs,价带最大值 (VBM) 位于 Y 点,而导带最小值 (CBM) 位于 Γ (图3a)。单层 SiAs 的间接带隙为 E g =1.72 eV 在 PBE 方案中。还可以看到 Y 处的 VBM 状态 点包括 p 是 轨道,而 Γ 的 CBM 点主要包括 s 轨道,这意味着外部变形将对两种状态产生不同的影响,并可能导致间接 - 直接转变,如下所示。与 SiAs 不同,单层 SiAs2 是一种几乎直接的半导体,VBM 位于 Y 的一侧 点和 CBM 有一点位移(图 3b)。 SiAs2 单层间接带隙为 E g =1.42 eV 在 PBE 方案中。 SiAs2 单层的 VBM 和 CBM 由 p 是 轨道和 s 轨道,分别。为了获得更准确的带隙值,我们还对 SiAs 和 SiAs2 单层进行了混合泛函计算 (HSE06)[46, 47]。从计算的能带结构(图 3a、b 的右侧)来看,PBE 和 HSE 的能带态锐度基本相同,在混合函数计算中仍然预测了间接带隙,但间隙值是SiAs和SiAs2分别增加到2.39 eV和2.07 eV。
<图片>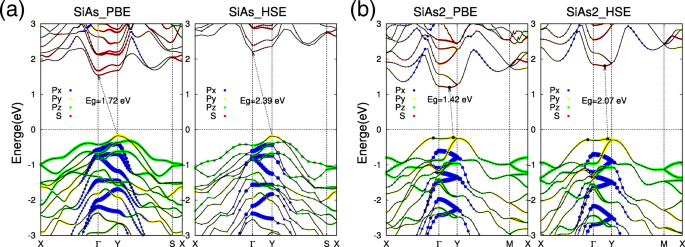
通过 PBE 和 HSE06 计算的单层 SiAs 和 SiAs2 的能带结构。单层SiAs和SiAs2能带结构的电子轨道分解表示为a 和 b , 分别。红点表示 s 轨道,而蓝色、黄色和绿色是 p x ,p 是 , 和 p z , 分别。费米能级设置为零并用虚线表示
载流子迁移率是新发现的二维材料在现代电子设备中潜在应用的关键因素,与 CBM 和 VBM 的带隙和位置一样重要。为了获得有关 SiAs 和 SiAs2 单层电子结构特性的更多细节,我们然后基于变形势 (DP) 理论计算了它们的声学声子限制载流子迁移率(包括 x 和 y 方向的电子和空穴)[48]在室温 (T =300 K )。在低能量状态 (300 K ),电子-声-声子散射在载流子传输中占主导地位,这使得声学声子限制成为预测许多二维结构载流子迁移率的有效方法,例如 MoS2 单层 [49]、碲烯 [19]、磷烯 [ 50] 和少层 MoO3 [51]。计算的有效质量m * 和载波移动μ SiAs 和 SiAs2 单层显示它们都具有高迁移率和传输各向异性(参见附加文件 1:表 S1 以及图 S3 和 S4),就像黑色磷烯 [50]。为了估计 SiAs 和 SiAs2 的载流子迁移率,我们首先使用近自由电子模型对它们的能带进行拟合,以获得有效载流子质量。对于 SiAs,我们定义 x 和 y 作为垂直于晶格向量的方向 b 和 a , 分别。沿x方向的\(m_{e}^{*}\)和\(m_{h}^{*}\)约为0.15 m 0 和 0.86 m 分别为 0,沿 y 方向为 0.80 m 0 和 0.22 m 0 (米 0 是自由电子质量),分别。对于 SiAs2,晶格矢量 a 的方向 定义为 x , 而 b 是 y .沿x方向的\(m_{e}^{*}\)和\(m_{h}^{*}\)约为0.14 m 0 和 0.65 m 分别为 0,沿 y 方向为 2.05 m 0 和 1.82 m 0,分别。我们进一步研究了弹性常数 (C) 和变形势 (E1)(参见附加文件 1:图 S2 和 S3)。基于上面得到的m * , C 和 E1 值,我们估算了载流子迁移率,如表 1 所示。 SiAs(SiAs2) 沿 x 的电子迁移率 和 y 方向为 0.66(0.26) 和 0.54(0.11) × 10 3 · 厘米 2 V −1 S −1 ,而沿 x 的空穴迁移率 和 y 方向为 3.90(0.13) 和 0.30(0.65) × 10 3 · 厘米 2 V −1 S −1 , 分别远高于 MoS2 [49]。
为了进一步阐明单层 SiAs 和 SiAs2 中 Si 和 As 原子的潜在键合机制,使用 PBE 功能的它们的总和部分态密度(PDOS),其电子密度分布对应于 VBM 和 CBM,在分别见图4。可以看到 As 和 Si 原子的 PDOS(图 4a,c)显示了 s 的强杂化 和 p 轨道,表明它们之间存在强共价键。单层SiAs和SiAs2的区别在于p的定位 z 轨道,这归因于 As 原子的不同键合配位环境。 SiAs 和 SiAs2 单层中位于 As 原子处的孤对电子态增加了三个最近的键合轨道,以决定单层结构屈曲的形成并形成 p z 轨道定位作用。在单层 SiAs 中,孤对通过 Si-As 键分开,这放松了排斥效应并拓宽了 p z 轨道。而在单层 SiAs2、As-As 键中,保留了 V 族半导体中非常常见的情况,将 p 局部化 z 轨道在更深的能级。
<图片>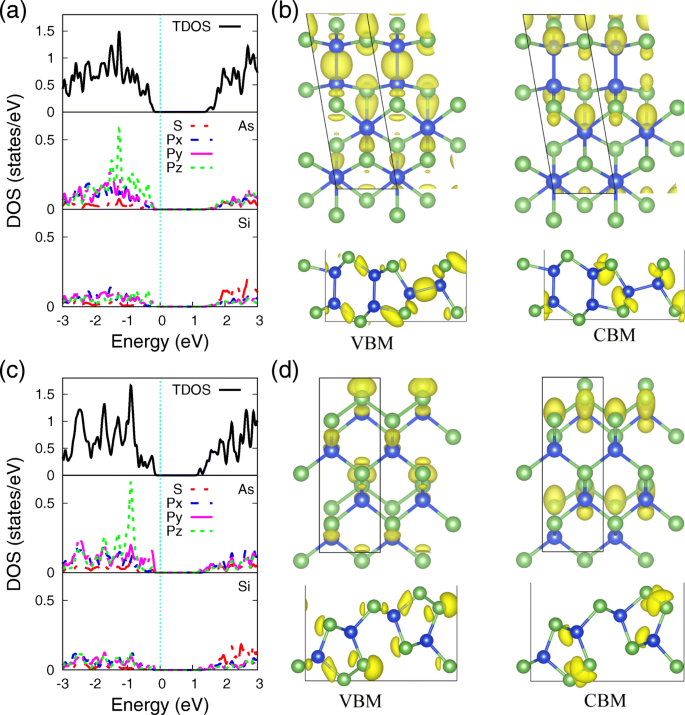
状态密度和 VBM 以及 CBM 的电子密度。 As 和 Si 原子的投影态密度 (PDOS) 以及 (a 的 VBM 和 CBM 对应的电子密度分布 , b ) SiAs 和 (c , d ) SiAs2 单层。等值面值 0.034 e /Å 3
正如我们所知,前沿态的特征不仅对微观理解传导通道感兴趣,而且对优化接触的设计也很重要。 [52]对应于单层 SiAs 和 SiAs2 的 VBM 和 CBM 的电荷密度分别显示在图 4b 和 d 中。 VBM 几乎是 Si 和 As 的 3p 轨道的杂化,而 CBM 主要来自 Si 和 As 的 3s 轨道的贡献,这也与图 4a、c 中的 PDOS 结果和能带结构的电子轨道分解一致图3。
机械应变是调节二维材料电子特性的有效方法,广泛用于改变黑色和蓝色磷烯和其他纳米片材料的能带结构 [53-55]。特别是,对于屈曲结构系统,能量成本通常很小,会引起显着的应变。在这里,通过在几何优化过程中改变每个原子的晶格常数和内部自由度来模拟机械应变的应用。应变 ε 定义为 ε =(l -l 0)/l 0,其中 l 和 l 0 是单层 SiAs 和 SiAs2 的应变和平衡晶格常数。在图 5a、b 中,分别表示了 2D SiAs 和 SiAs2 在应变下的高屈曲几何结构的详细变化。可以看到,通过改变屈曲角度θ,它们的屈曲高度被扩大或压缩 1(2) 具有近乎线性变化的双轴压缩或拉伸应变。我们还发现它们的高屈曲几何结构在相当大的应变下仍然保持良好,其声子光谱,如附加文件 1:图 S5 和 S6 所示,即使在大应变范围内也不存在负频率。双轴压缩和拉伸应变下单层 SiAs 和 SiAs2 的间隙变化分别如图 5c、d 所示。可以看出,SiAs和SiAs2的电子性质对应变敏感,在一定应变区经历了间接到直接的能带跃迁,然后在大应变区发生了金属跃迁。
<图片>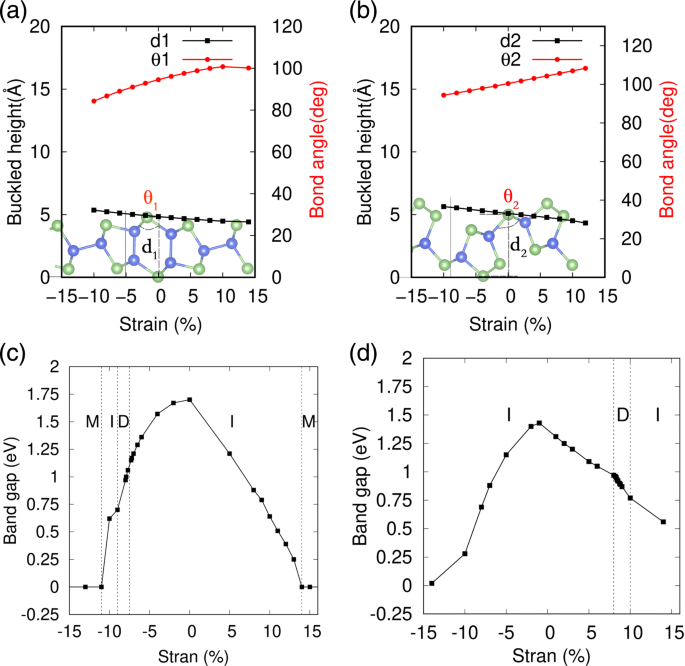
应变对二维 SiAs 和 SiAs2 的几何结构和带隙的影响。 一 , c 代表SiAs;和 b , d 表示 SiAs2; M、I、D分别代表金属、间接半导体和直接半导体
SiAs 和 SiAs2 能带结构的详细变化如图 1 和图 2 所示。分别为 6 和 7。在双轴压应变下,单层 SiAs 的屈曲高度增加,煤层气从 Γ 偏移 到 Y-S 线上的一点,然后回到 Y。而 VBM 保持在 Y 点静止,直到压缩应变达到 ε =- 10% .因此,随着压缩应变的增加,带隙从间接 Y 切换到 Γ , 通过间接 Y 到 Y–S 线上的一个点,将 Y 引导到 Y 并返回到 Γ 上的间接点 -Y 线到 Y,如图 6 所示。对于拉伸应变,Y 处的 VBM 移动到 Y-S 线上的一点,而 CBM 位于 Γ 移动到 Y 并且带隙保持间接。对于大应变,无论是压缩还是拉伸都会导致金属转变,如图 5c 所示。
<图片>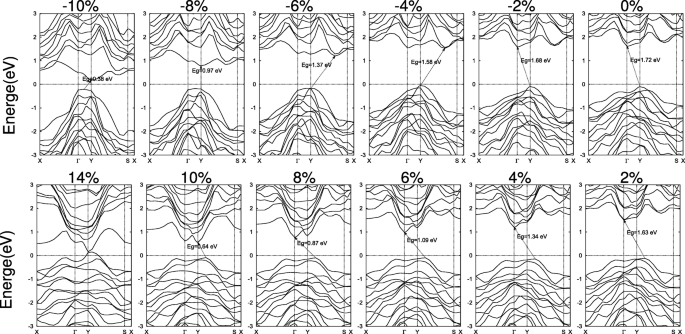
双轴应变下二维 SiAs 的能带结构。费米能级设置为零并用虚线表示
<图片>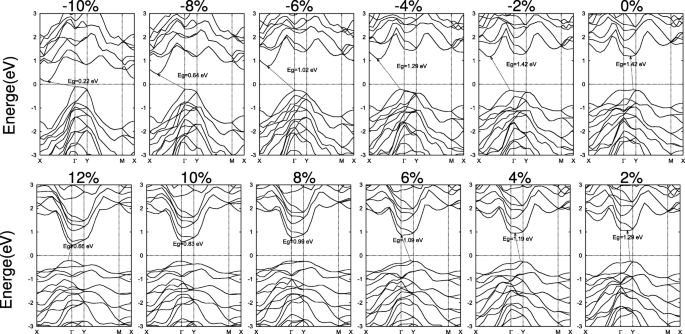
双轴应变下二维 SiAs2 的能带结构。费米能级设置为零并用虚线表示
在图 7 中,对 2D SiAs2 进行了类似的研究。 8-10% 范围内的拉伸应变不是压缩,而是导致直接带隙。当单层 SiAs2 在拉伸应变下随着屈曲高度的减小而扩展时,VBM 从 Γ 上的一个点偏移 -Y线到Γ 并保持在 8-10% 的范围内,然后移开到 Γ 上的一个点 –X 线,而 CBM 从 Γ 上的一点移动 -Y线到Γ 并坚持下去。因此,随着拉伸应变的增加,带隙从 Γ 上的间接转换 -Y 线指向 Γ – Γ 然后回到 Γ 上的间接点 –X线到Γ ,如图 7 所示。压缩应变仍然是间接带隙。并且大应变具有类似的作用,导致金属转变为 SiAs。
应变 SiAs 和 SiAs2 的代表性直接能带结构也显示在附加文件 1 中:图 S7a 和 b 通过 PBE 和 HSE 计算。对于 SiAs,E 的直接带隙 g =1.75 eV (HSE) 将 VBM 和 CBM 定位在 Y 点是在 ε 的双轴压缩应变下获得的 =- 7.5% .与 SiAs 不同,ε 的双轴拉伸应变 =8.5% 将 SiAs2 诱导到 E 的直接带 g =1.60 eV (HSE)。 VBM 和 CBM 位于 Γ 点。
结论
总之,通过第一性原理 DFT 计算,我们提出了两种新的硅和砷化合物二维材料 SiAs 和 SiAs2,它们在动态和热力学上都是稳定的。我们的计算表明 SiAs 和 SiAs2 单层是带隙为 2.39 eV 的间接半导体 和 2.07 eV , 分别。 SiAs 和 SiAs2 单层的带隙对应变敏感,在一定的机械应变下,它们经历间接到直接的能带跃迁,甚至对金属。 SiAs 和 SiAs2 单层比 MoS2 具有更高的迁移率,并显示出像黑色磷烯一样的各向异性传输。我们的工作为纳米级光学器件的新功能开辟了一条新途径。
缩写
- 二维:
-
二维
- CASTEP:
-
剑桥序贯总能量包
- CBM:
-
导带最小值
- DFT:
-
密度泛函理论
- DFPT:
-
密度泛函摄动理论
- DP:
-
变形势
- GGA:
-
广义梯度逼近
- MD:
-
分子动力学
- NVT:
-
摩尔-体积-温度
- 爪子:
-
投影仪增强波
- PBE:
-
Perdew-Burke-Ernzerhof
- PDOS:
-
偏态密度
- TMD:
-
过渡金属二硫属化物
- VASP:
-
Vienna ab initio 仿真包
- VBM:
-
价带最大值
纳米材料
- 使用来自轧制废料的磁铁矿纳米吸附剂从水溶液中吸附去除铜 (II) 离子:合成、表征、吸附和动力学建模研究
- 超窄带完美吸收体及其在可见光区域中作为等离子体传感器的应用
- 硼烯稳定性和STM图像的第一性原理研究
- RGO 和三维石墨烯网络共同修饰的高性能 TIM
- 钯(II)离子印迹聚合物纳米球的制备及其从水溶液中去除钯(II)
- 5-氨基乙酰丙酸-角鲨烯纳米组件用于肿瘤光检测和治疗:体外研究
- GaAs/AlAs 超晶格点缺陷的第一性原理研究:相稳定性以及对能带结构和载流子迁移率的影响
- GaTe/C2N 异质结构中的应变可调电子特性和能带排列:第一性原理计算
- 研究二硫化钼和 ZrO2 异质结处的能带
- 来自第一性原理研究的 SiAs 和 SiAs2 单层中的应变可调带隙和高载流子迁移率
- 空心聚苯胺微/纳米球的制备及其从废水中去除 Cr (VI) 的能力
- 氢化石墨烯/六方氮化硼异双层载流子迁移率的理论研究