

由胶体纳米纤维素 (CeO2 - x) 中的氧扩散驱动的铈氧化状态的振荡
摘要
CeO2 的氧化还原性能 - x 纳米晶体(nanoceria)总是伴随着 Ce 3+ 之间铈氧化态的转换 和 Ce 4+ .我们监测了 Ce 3+ → Ce 4+ 胶体水溶液中氧化剂刺激纳米氧化铈的氧化控制Ce 3+ 的发光 离纳米氧化铈表面不同距离的离子。观测到的 Ce 3+ 发光变化表明Ce 3+ → Ce 4+ 反应在纳米氧化铈内部发生,由来自氧化纳米氧化铈表面上的水分解产生的扩散氧触发。我们首次观察到 Ce 3+ 的明显振荡 Ce 3+ 产生的发光强度 ↔ Ce 4+ 可逆切换。当纳米氧化铈晶格中的氧空位浓度、胶体溶液中的氧化剂浓度和温度达到一定的临界值时,这种阈值效应是由纳米氧化铈吸收和释放氧来驱动的。因此,纳米氧化铈根据环境氧化还原条件吸收和释放氧气的能力确实使其成为自给自足的永恒抗氧化剂。
背景
今天,具有不同结构和化学成分的纳米晶体被广泛用于现代应用的多样性 [1,2,3,4,5,6,7,8,9]。随着重要的工程应用 [3, 4],CeO2 纳米晶体(nanoceria)催生了有希望的生物医学发展 [5,6,7,8,9],因为它能够作为活性氧 (ROS) 的再生清除剂.使纳米氧化铈如此独特和有用的主要先决条件通常归因于氧空位 (VO) 和 Ce 3+ 的高含量 其表面的离子 [10,11,12,13,14]。在纳米氧化铈晶格中,VO 和 Ce 3+ 离子是相互关联的缺陷 [10,11,12,13,14];两个Ce 3+ 离子占一个 VO [13]。缺陷 (Ce 3+ ,VO) 在纳米氧化铈中的浓度可以通过粒径、特殊掺杂和温度处理来控制 [11, 14, 15]。一般来说,表面氧可以通过 VO 的产生和愈合来辅助氧化还原循环,或者表面 VO 可以作为催化活性物质的结合位点 [3, 4, 14]。表面Ce 3+ 由于在 3+ 和 4+ 氧化态之间切换,纳米氧化铈离子通常被认为提供 ROS 清除 [5,6,7,8,9]。低浓度的 ROS,即超氧离子 \( {\mathrm{O}}_2^{-} \)、羟基自由基 OH˙ 和过氧化氢 H2O2,对于细胞功能的调节至关重要 [5,6,7 ,8,9]。与在与 ROS 相互作用后不可挽回地消失的普通抗氧化剂 [5,6,7] 不同,纳米氧化铈的粒径低于 15 nm,可以作为一种自我再生的抗氧化剂 [5,6,7,8,9]。纳米氧化铈生物活性对其大小的关键依赖性,以及纳米氧化铈在生物环境中的自我再生机制,仍然知之甚少 [5,6,7],讨论仍在继续 [8, 9]。应该强调的是,在体外和体内实验 [5,6,7,8,9] 中,纳米氧化铈在高缺陷浓度和水活性下运行,其氧化还原性能会被细胞抗氧化系统强烈掩盖。
因此,为了了解纳米氧化还原性能的机制,我们使用了更简单和可控的条件:在含氧量变化的纳米氧化铈样品的胶体水溶液中研究了纳米氧化动力学。作为 VO (Ce 3+ ) 浓度随颗粒大小而变化 [10,11,12],使用了 3.0、10.0 和 50.0 nm 的纳米氧化铈样品。根据数据[12],3.0-nm纳米氧化铈中的VO浓度可达~ 20%。我们确定 10.0 nm 纳米氧化铈中的 VO 浓度比 3.0 nm 纳米氧化铈低两倍(参见附加文件 1)。在 50.0-nm 纳米氧化铈的情况下,掺杂 Y 3+ (或 Eu 3+ ) 离子和真空退火用于产生 VO 并为纳米氧化铈晶格中的氧扩散创造不同的条件 [14, 15]。在所有 50.0 nm 样品中,VO 浓度与 10.0 nm 纳米氧化铈相等。在 Re 3+ 掺杂50.0-nm纳米氧化铈,Y 3+ 的浓度 和 Eu 3+ 离子处于 10 原子百分比的水平(参见附加文件 1)。这些浓度远低于这些离子在二氧化铈晶格中的相应溶解度极限值,~ 25 at.% [16] 或什至~ 45 at.% [17] Y 3+ 离子和 ~ 30 at.% [18] 对于 Eu 3+ 离子;因此,可以排除 Y2O3 或 Eu2O3 相的形成。所有胶体溶液都含有相同量的物质 (1.0 g/l),并以初始 pH 值~ 7 为特征。对于纳米氧化铈,使用过氧化氢 (HP) 和高碘酸钾 KIO4 (PP)。 PP 允许我们排除 HP 的化学多样性。获得的样品的纳米氧化铈合成和表征的详细信息,以及实验的描述,在附加文件 1 中提供。
结果与讨论
依靠我们的初步实验[19],Ce 3+ 由于 Ce 3+ 的偶极子允许 5d → 4f 光学跃迁导致的发光(图 1a) 离子 [20] 用于监测所有测试纳米氧化铈样品的氧化动力学。 Ce 3+ 的增加 随着粒径的减小,即随着表面积与体积比的增加(参见图 1a 中的插图),谱带不对称清楚地表明其不均匀性。该波段的长波部分可归因于 Ce 3+ 来自纳米氧化铈次表层的发光,以及剩余部分的 Ce 3+ 带来自深层次的Ce 3+ 离子。地下Ce 3+ 离子具有红移的发光光谱(参见图 1a 中的插图),因为晶格参数在纳米氧化铈表面的方向上增加导致作用在这些离子上的晶体场减弱 [12]。与 3.0-nm 纳米二氧化硅相反,对于 50.0-nm 纳米二氧化硅,这些离子对所得发光光谱的影响可以忽略不计(参见图 1a 中的插图)。 Ce 3+ 长波部分更强的 Forster 猝灭 [21] 证实了这一归因 带(参见附加文件 1 和图 1b)。淬灭剂浓度的增加导致深部Ce 3+ 的发光猝灭 离子(图 1b),因为供体 - 受体距离的减少 [21]。
<图片>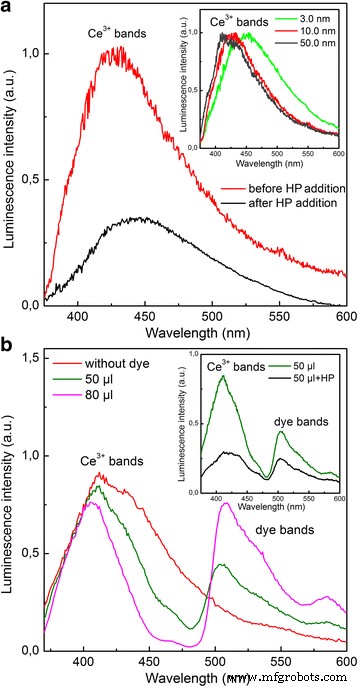
纳米氧化铈在不同条件下的发光光谱。 一 HP (C =0.1 mM)添加。插入:不同纳米氧化铈样品的归一化光谱。 b 添加 50 和 80 微升染料之前和之后的 10.0 纳米纳米氧化铈的发光光谱。插入:加入 50 μl 染料并随后加入 HP (C =0.1 mM)
在胶体溶液中加入氧化剂(HP 或 PP)导致 Ce 3+ 所有测试纳米氧化铈样品的条带强度(图 1a)。此外,可以看出,在选择性淬灭 Ce 3+ 带(图 1b),氧化剂刺激了深层 Ce 3+ 的发光下降 离子(参见图 1b 中的插图)。 Ce 3+ → Ce 4+ 尽管氧化剂不能渗透到纳米氧化铈中,但这些离子也会发生氧化。这种效果类似于在氧气气氛中对纳米氧化铈进行退火(参见附加文件 1)。因此,氧化剂刺激氧(其来源将在下面确定)渗透到纳米氧化铈内。 Ce 3+ 的缺陷控制时间演化确实证实了这一点 在氧化作用下发光(图 2a)。在这个实验中,HP 和 PP 都以类似的方式起作用(见图 2a 中的插图)。从图 2a 可以看出,具有最高 VO 浓度的 3.0 nm 纳米氧化铈表现出最快的 Ce 3+ 带强度和最低残留 Ce 3+ 发光。在相同的 VO 浓度下,Y 3+ -掺杂的纳米氧化铈表现出更强的Ce 3+ 淬火速率 带强度和较低水平的残留 Ce 3+ 与 Eu 3+ 相比的发光 掺杂的纳米氧化铈(图 2a)。这与Eu 3+ 存在时氧化铈中氧扩散的活化能增加有关 比 Y 3+ 更强的离子 离子 [14, 15]。因此,Ce 3+ 氧化作用下的谱带强度降低(图 1 和 2a)是 Ce 3+ 的结果 → Ce 4+ 氧通过扩散的空位机制渗透到纳米氧化铈中引起的氧化 [14, 15]。在纳米氧化铈氧化时向胶体溶液中加入还原剂(例如苯三醇)不会导致 Ce 3+ 发光恢复,这与所提出的 Ce 3+ 机制一致 → Ce 4+ 纳米氧化铁内部氧化。
<图片>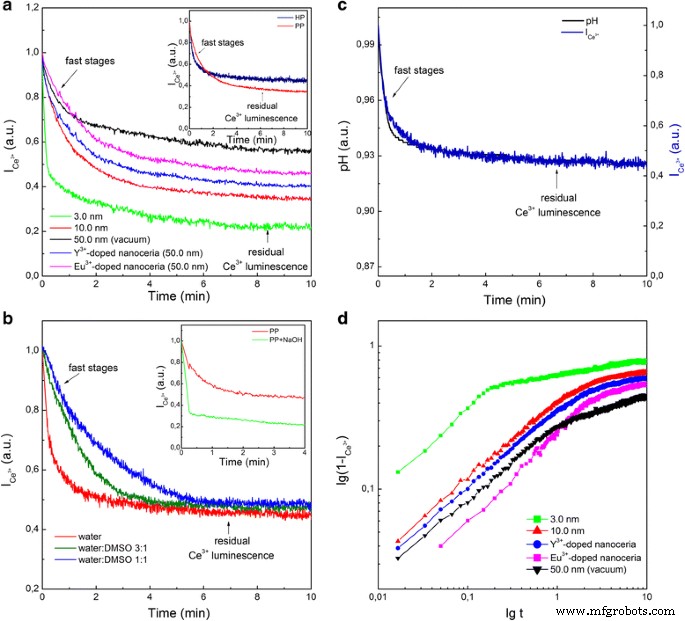
Ce 3+ 的时间演化 在氧化作用下发光(C =0.1 mM) 用于纳米氧化铈的不同样本。 一 HP加成后;插入——在添加 HP 和 PP 之后; b 在水-DMSO 溶液中将 PP 添加到 10.0 nm 纳米氧化铈后;插入——在加入 PP 之后以及在加入 NaOH 和随后的 PP 之后。 c pH 和 Ce 3+ 添加 10.0 nm 纳米氧化铈的 PP 后的带强度 (\( {I}_{{\mathrm{Ce}}^{3+}} \))。 d 氧ρ填充纳米氧化铈曲线 (t )=1−\( {I}_{{\mathrm{Ce}}^{3+}}(t) \) 使用 a 中显示的数据显示在对数图上
胶体溶液中较低的水浓度减缓了纳米氧化铈的氧化动力学(图 2b),而初始 pH 值的增加加速了这一过程(参见图 2b 中的插页)。排除可能的 H + 来源 在 HP 应用的情况下,我们还揭示了 pH 值降低与 Ce 3+ PP 在纳米氧化铈下的谱带强度下降(图 2c)。这些事实表明,在Ce 4+ 的参与下可以高效地进行水分解 \( \hbox{--} {\mathrm{V}}_{\mathrm{O}}^{++} \)–Ce 4+ (或 Ce 4+ \( \hbox{--} {\mathrm{V}}_{\mathrm{O}}^{+} \)–Ce 3+ ) 由于 Ce 3+ 氧化而在纳米氧化铈表面形成的活性位点 -VO-Ce 3+ 站点(图 3)[13]。有两种可能的方法(图 3):O 2− 离子占据 \( {\mathrm{V}}_{\mathrm{O}}^{++} \) 和两个 H + 离子被喷射到溶液或 O 2− 离子占据 \( {\mathrm{V}}_{\mathrm{O}}^{+} \) 导致产生羟基,这使得一个 H + 离子被喷射到溶液中(图 3)。氧气第一次跃入纳米氧化铈会再生 Ce 3+ –VO–Ce 3+ 新氧化循环的位置(图 3)。该过程可以根据氧化剂浓度以不同的速率重复多次。曲线 \( \uprho (t)=1-{I}_{{\mathrm{Ce}}^{3+}}(t) \) 描述了纳米氧化铈的充氧过程,其初始阶段与~t 1/2 功能(见图 2d)。这意味着氧气通过单列扩散通过 VO 通道(图 3)渗透到纳米氧化铈中,其中氧原子不能相互绕过 [22, 23]。由于两个原因,在纳米氧化铈的氧终止平面上形成大的 VO 簇(图 3)是不可避免的:所有测试的纳米氧化铈样品都含有足够高的 VO 浓度接近渗透阈值 [24] 并且 VO 浓度达到其最大值靠近纳米氧化铈表面 [10,11,12]。观察到的地下 VO 的线性结构 [25, 26] 可以被认为是 VO 通道或大 VO 簇的组成部分。
<图片>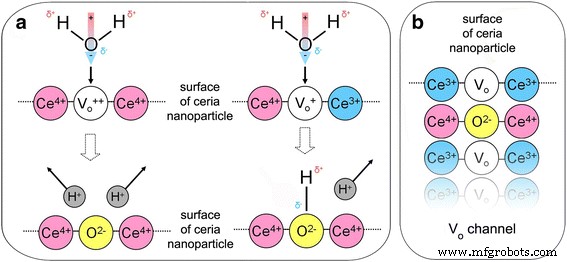
纳米氧化铈与氧化剂和水分子相互作用的阶段。 一 双氧化Ce 4+ –\( {\mathrm{V}}_{\mathrm{O}}^{++} \)–Ce 4+ 位点和单氧化 Ce 4+ –\( {\mathrm{V}}_{\mathrm{O}}^{+} \)–Ce 3+ 纳米氧化铈表面的位点及其与 H2O 分子的相互作用。 b Ce 3+ 的再生 –VO–Ce 3+ 下一个氧化循环的场所
Ce 3+ 的明显振荡 当胶体溶液中的氧化剂(HP 或 PP)浓度超过 ~ 0.5 mM 时观察到条带强度,因此纳米氧化铈 Ce 3+ 的动力学 → Ce 4+ 氧化转化为Ce 3+ ↔ Ce 4+ 氧化还原场景(图 4)。这些振荡在 HP (C =1.0 mM) 或 PP (C =1.0 mM)添加,但在快速阶段完成时开始发展(图 4a、b)。对于所有氧化剂,在振荡消失后,可以通过向胶体溶液中添加新的氧化剂部分来重复它们(图 4c)。为了比较,Ce 3+ 的振荡 所有测试的纳米氧化铈样品的条带强度如图 4d 所示。图 4d 中的基线实际上是残差 Ce 3+ 的水平 每个纳米氧化铈样品的发光(图 2a),以及 Ce 3+ 波段强度在这些水平之上振荡。 Y 3+ 中VO浓度的变化 (或 Eu 3+ ) 掺杂的 50.0-nm 纳米氧化铈清楚地表明 Ce 3+ 只有当 VO 浓度与 10.0 nm 纳米氧化铈中的浓度相等时,才能观察到带强度(图 4d)。在 Eu 3+ 的情况下 掺杂的 50.0 nm 纳米氧化铈,振荡更不规则且不那么明显(图 4d)。正如前面提到的,这个事实与在 Eu 3+ 存在下抑制氧扩散是一致的 离子 [14, 15]。在具有热力学非平衡 VO 的退火 50.0 nm 纳米氧化铈中,根本没有观察到振荡。仅在 35°C 以上的温度下才能观察到振荡(见图 4)。
<图片>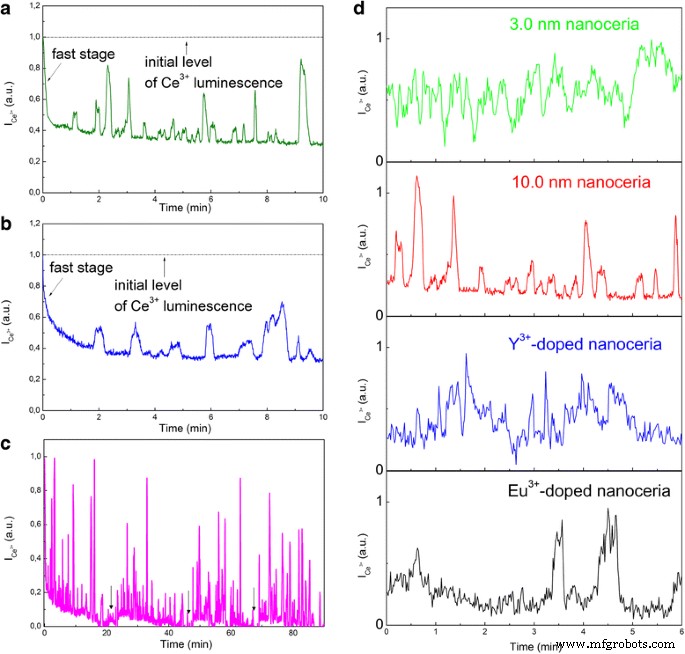
Ce 3+ 的振荡 带强度 (\( {I}_{{\mathrm{Ce}}^{3+}} \)) 由氧化剂 (C =1.0 mM) 在胶体纳米氧化铈中:a 10.0-nm 纳米氧化铈的 HP 添加后; b 为 10.0 nm 纳米氧化铈添加 PP 后; c 对 10.0-nm 纳米氧化铈添加多次 HP 后; d 不同纳米氧化铈样品添加HP后
观察到的振荡(图 4)出现在快速阶段(图 2a 和 4a)期间由氧气积累的 VO 数量变得强烈不平衡时。第一个振荡峰值的增长(图 4a、b)伴随着过量氧的释放,只要观察到振荡,纳米氧化铈就会释放(Ce 3+ 发光增加)和吸收(Ce 3+ 发光减少)氧气。对于退火的 50.0 nm 纳米氧化铈,氧释放阶段,因此,Ce 4+ ↔ Ce 3+ 振荡是不可能的,因为热力学非平衡 VO 被氧化过程中积累的氧气不可逆地修复(图 2a 和 4)。
需要注意的是,Ce 3+ 的时间尺度 带强度演化(见图 2 和 4)需要在室温下纳米氧化铈中异常高的氧扩散速率。通常,在氧化物中,由于活化能很大,普通的氧扩散太慢[14, 15]。但在我们的例子中,快速氧扩散是由单列扩散固有的活化能的负载依赖性降低提供的 [23]。胶体溶液中的氧化剂浓度通过氧填充VO簇的速率和水平来控制这种效应。
结论
我们的结果表明了纳米氧化还原性能背后的微观机制的新视野。首先,既表面Ce 3+ 可用于氧化剂和深层Ce 3+ 的离子 由于开放的 VO 簇支持氧扩散,离子参与纳米氧化铈的氧化动力学。这种 VO 簇不可避免地在纳米氧化铈的足够小尺寸(<15 nm)处形成,这解释了纳米氧化铈抗氧化活性的强尺寸依赖性。自我再生(反向Ce 4+ → Ce 3+ 纳米氧化铈在生物环境中的还原)是通过 ROS 释放其氧化过程中积累的氧气的结果。类似于多相催化中的振荡[27],纳米氧化铈中铈氧化态的振荡可用于开发高性能抗氧化剂,这对于高强度辐射下的细胞保护(癌症放射治疗、核灾难等)。总体而言,论文中提出的想法使人们开始理性地寻找新的纳米材料,这些材料不仅可以用作有效的抗氧化剂,还可以用作各种技术领域的独特催化材料。
方法
纳米纤维素合成方法
3.0 和 10.0-nm Nanoceria 的胶体合成
通过以下方法获得二氧化铈纳米颗粒的水溶液:将CeCl 3 溶液(100ml,2mM)与100ml六亚甲基四胺溶液(4mM)混合并在室温下借助磁力搅拌器搅拌3小时。之后,将 1.8 ml NH4OH 和 0.6 ml H2O2 添加到溶液中。然后,将溶液放入圆底烧瓶中并回流 5 小时。结果,获得了透明的无色溶液。将溶液在旋转蒸发器烧瓶中在真空下在 70°C 的浴温下蒸发至 30ml。将 2 M NaCl 溶液加入到所得溶液中直至所得溶液变浑浊。然后,通过离心沉淀固相。分离沉淀,再次加入氯化钠溶液。沉淀清洗过程重复3次。在离心的最后阶段之后,将摩尔比为 1:1 的 CeO2/NaCt 的柠檬酸钠溶液加入到沉淀物中。从摩尔比为 1:10 的氯化铈 (III) 和六亚甲基四胺 (HMTA) 的混合物获得的纳米氧化铈的尺寸为~ 10.0 nm。随着 HMTA 的进一步增加,获得的纳米颗粒的过大尺寸减小到~ 3.0 nm。 СеО2 - х 纳米粒子由摩尔比为 1:1 的柠檬酸钠稳定。将溶液另外用去离子水透析 24 小时以去除过量的离子和有机物种类。使用了截留分子量为“Cellu Sep H1”3.5 KDa 的透析膜管。所有溶胶在透射光下都是透明的,并通过孔径为100 nm的膜过滤器而没有损失。
50.0-nm Nanoceria 的溶胶-凝胶合成
СеО2 - x , СеО2:Eu 3+ /Y 3+ (0.1–10 at.%) 纳米晶体是通过 Pechini 方法获得的。氧化铈 (СеO2) (99.995%, Sigma-Aldrich) 在 60 °C 下溶解在硝酸 (НNO3) 和过氧化氢 (Н2О2)(体积比为 1:1)的混合物中。在 80°C 下,将氧化铕 (Eu2O3)(99.999%,Sigma-Aldrich)和氧化钇 (Y2O3)(99.999%,Sigma-Aldrich)溶解在稀释的 НNO3 中。将 0.75 克柠檬酸和 1 毫升乙二醇的溶液加入 20 毫升硝酸铈 Се(NO3)3 (C =1 M) 溶液或 20 毫升硝酸铈 Се(NO3)3 (C =1 M) 和硝酸铕 Eu(NO3)3/硝酸钇 Y(NO3)3 (C =1 M) 解决方案。所有得到的混合物在 80°C 下处理 10 小时,然后通过 10 质量%的 NH3 水溶液水解。沉淀物在 120°C 下干燥 5 小时,然后在 250°C 下脱水 4 小时。纳米晶体在真空中在 1000°C 下退火 2 小时。退火后,纳米颗粒以 1 g/l 的浓度分散在水中。
实验技巧
连续波 GKL-4UM He-Cd 激光器 (λ ~325 nm) 并使用 SDL-1 光栅单色器和 Hamamatsu R9110 PMT 在光子计数模式下注册。
在纳米氧化铈胶体水溶液中加入氧化剂(过氧化氢或高碘酸钾)后,Ce 3+ 的时间演变 发光强度(在 390 nm 处拍摄)是通过 CW 激发(He-Cd 激光)下的时间分辨测量来确定的。水溶液中纳米氧化铈的浓度在所有实验中都相似并且等于 1 g/l。引发不可逆 Ce 3+ 所需的氧化剂浓度 → Ce 4+ 氧化还原反应等于 0.1 mM;引发可逆 Ce 3+ 的氧化剂浓度 ↔ Ce 4+ H2O2 (HP) 和 KIO4 (PP) 的氧化还原反应均等于 1.0 mM。
使用pH计测量添加氧化剂后纳米氧化铈胶体水溶液的pH值的时间依赖性。作为氧化剂 HP (C =0.1 mM) 和 PP (C 0.1 mM),在将部分氧化剂加入纳米氧化铈胶体水溶液后,以 1 秒的时间间隔记录 pH 值。以加入氧化剂后蒸馏水pH值的时间依赖性为对照(蒸馏水的初始pH值与纳米氧化铈胶体溶液的pH值相同(pH =7)。
所有实验均在 T 实现 =37°С。
缩写
- HP:
-
过氧化氢
- PP:
-
高碘酸钾
- ROS:
-
活性氧
- 画外音:
-
氧空位
纳米材料