

Al-Doped BiFeO3 粉末局部结构的同步加速器 X 射线吸收光谱研究
摘要
Al掺杂的BiFeO3,即BFAx 带有 x 的 O 粉末样品 =0、0.025、0.05 和 0.1,通过水热途径制备。 Al取代对BFAx结构、电学和光学性能的影响 研究了 O 样品。发现在BiFeO3的B位取代Al离子没有引起结构变化,仍然保留了带有R3c的菱形钙钛矿结构 X 射线衍射 (XRD) 和拉曼测量证实了对称性。 Fe K 上方的 X 射线吸收精细结构 (XAFS) -edge 和 Bi L BFAx 中的 3 边 还测量和分析了 O 粉末。 Fe 离子表现出混合价态 (Fe 2+ /Fe 3+ ) 而 Bi 离子在所有样品中保持 + 3 价态。铁 K -edge XAFS 也表明 Fe 3d 的杂交之间存在竞争 和 Al 3d 与 O 2p 轨道和更多 4p 的出现 Al掺杂的轨道。 Bi L 3-edge XAFS 揭示了从 2p 的过渡 3/2 到 6d 状态增加,所以 6d 的能量也增加 状态。此外,Al 离子掺杂影响 Fe 原子的最近邻和次最近配位壳和 Bi 原子的最近邻壳。紫外-可见 (UV-Vis) 光谱结果显示 BFAx 水热法制备的O可能是一种合适的可见光光催化材料。
背景
多铁性材料是同时具有铁电性、铁磁性和铁弹性等铁性特性的材料[1]。这种材料表现出有趣的行为,例如电极化,可以通过施加外部电场来控制,反之亦然。对这些材料的兴趣是由于它们在化学生物传感器、纳米电子和高密度数据存储设备等方面的广泛应用 [2, 3]。钙钛矿结构具有通式 ABO3,其中 O 是离子(通常指氧),A 和 B 分别是阳离子。一般来说,A位阳离子较大,价态较低,与O 2- 结合 形成密堆积层(即,在晶胞的角部)。 B位阳离子较小,价态较高,可用于氧八面体配位环境(即氧阴离子八面体的中心)[4]。根据定义,几乎所有多铁性材料都是具有低转变温度的反铁磁 (AFM) 或弱铁磁 (FM)。它们分为两类:单相和复合。然而,自然界中很少发现同时具有铁电 (FE) 和 FM 特性的单相多铁性材料 [5]。在当今已知的所有多铁性材料中,铁酸铋 (BiFeO3; BFO) 是具有菱形畸变钙钛矿晶格类型的单相材料之一,具有极性空间群 R3c . BFO 显示居里温度为 T 的 FE C ≈ 1103 k 和 G 型 AFM 排序,尼尔温度为 T N ≈ 643 k,高于室温 (RT) [6]。这种材料沿 [111]c 表现出 AFM G 型自旋构型 或 [001]h 在其伪立方体或菱面体结构中的方向,并具有叠加的螺旋自旋结构,沿 [110]h 的周期约为 62 Å 轴在 RT [7]。它具有约 90 μC/cm 2 的大本征自发极化 归因于八面体 FeO6 的畸变,由于 6 s 的存在 2 孤对电子[8]。此外,FE 和磁序参数之间存在磁电耦合。 FE 特性取决于孤对电子,而 FM 特性取决于部分填充的内壳,即极化来自双位(A 位),而磁化来自 Fe 位(B 位)。
除此之外,不幸的是,由于铋和氧空位、杂质相、铁价波动和界面质量差等电荷缺陷,BFO 的主要缺点是电阻率低或漏电流大[9]。此外,由于Bi2Fe4O9(空间群Pbam ) 和 Bi25FeO39(空间群 I23 )。在制备过程中不可避免地会产生杂质。为了解决这些问题和局限性,几个研究小组一直在使用各种方法来克服 BFO 的缺陷,例如应变改性、二价和稀土离子掺杂的替代。现在,在该研究领域内,通过在 A 位或 B 位掺杂稀土元素或过渡金属离子,或在 A 和 B 位共掺杂,可以增强 BFO 的多铁性。例如,掺杂稀土元素可以稳定钙钛矿结构,保持非中心对称性,控制Bi 3+ 的蒸发 离子 [10]。掺杂过渡金属离子可降低Fe 3+ 的价态波动 离子。诸如用于 A 位取代的 Pr、Sm、Eu、Gd 和 La [11, 12] 以及用于 B 位取代的 Mn、Cr 和 Ti [13,14,15] 等元素已经被报道。此外,共掺杂可以增强磁性、介电性和铁电性。对于 BFO 的 A 位和 B 位的共掺杂,已经报道了 La-Gd、Ba-Ni、Dy-Cr、Y-Mn 和 Tb-Ti [16,17,18,19,20]。迄今为止,溶胶-凝胶法[21]、机械化学法[22]、自燃法[23]、脉冲激光沉积法[24]、等多种途径 据报道,热液和热液 [25, 26] 可以制备 BFO。水热法因其节能、分散细、成本低、粒径小等优点而得到广泛应用[27]。反应温度应足够高以形成 BFO,并在样品制备过程中用于去除第二相。 Azam 等人 [28] 已经研究了大多数先前在 BFO 的 A 位和 B 位掺杂的铝的结构、光学和传输特性。马杜等人。 [29] 报道了 B 位铝掺杂 BFO 的光催化应用。 Jawad 等人的另一份报告。 [30] 探索了纳米结构 BFO 陶瓷的介电行为。王等人的工作。 [31]详细研究了Al掺杂BFO的空心晶体。然而,一些重要的物理性质仍缺乏了解,例如 B 位掺杂对材料局部电子结构的影响。现阶段,X射线吸收精细结构光谱(XAFS)是研究原子局部环境的有力方法之一,提供材料的结构信息,以及吸收能、元素价态、电荷转移、和粘合类型[32]。据我们所知,目前还没有关于 B 位 Al 掺杂对 XAFS 研究 BFO 局部电子结构影响的报道。
在这项工作中,未掺杂的 BFO 和目标成分 BiFe1-x 铝x O3 (BFAx O) 与 x =0、0.025、0.05 和 0.1 是通过水热途径合成的。主要重点是研究在 B 位掺杂 Al 对 BFO 性能的影响,并与未掺杂的 BFO 进行比较。对其结构特性进行了详细研究。
方法
水热法制备未掺杂的BFO和BFAx O 样品。这项工作中使用的化学试剂是硝酸铋 (Bi(NO3)3·5H2O)、硝酸铁 (Fe(NO3)3·9H2O)、硝酸铝 (Al(NO3)3·6H2O) 和氢氧化钾 (KOH)。所有化学试剂均按原样使用,无需进一步纯化。 Bi(NO3)3·5H2O和Fe(NO3)3·9H2O为原料,Al(NO3)3·6H2O和KOH为添加剂。去离子水用于制备所有水溶液。制备 BFO 粉末的典型运行如下:将 Bi(NO3)3·5H2O、Fe(NO3)3·9H2O 和 Al(NO3)3·6H2O 各 20 mL 放入 80mL 不锈钢高压釜中和混合好。之后,将适量的KOH溶液缓慢滴加到之前的混合溶液中,直至充满其体积的65-80%,然后将其转移到强磁力搅拌装置中,在80 ℃下搅拌2-3 h至得到明确的解决方案。根据方法程序,将得到的深棕色溶液转移到衬有聚四氟乙烯的不锈钢高压釜中。水热处理在200 °C的温度下在自生压力下进行10 小时。加热速率为 2 °C/min。水热反应完成后,自然冷却至室温。随后,收集所得粉末并用丙酮、去离子水和乙醇洗涤数次,直至溶液的pH值达到7。最后,BFAx 将 O 粉末置于恒温干燥箱中,在 70 °C 下干燥 6 小时,然后干燥以进行进一步表征。我们准备了四组 BFAx 样本 通过改变 Al(NO3)3·6H2O 的浓度从 0-0.1 M 改变 O。
BFAx的晶体结构 O 样品通过 X 射线衍射(XRD,Mac Science M18XHF22-SRA)测定。使用来自 Ar+ 激光辐射的拉曼光谱 (Renishaw InVia Reflex) 来确定粉末在室温下的结构特性。 XAFS 数据是在中国北京同步辐射装置 (BSRF) 的光束线 1W2B 处以不同浓度以透射模式收集的。铁 K -边缘光谱,能量分辨率为 ΔE /E :2 × 10 −4 和 Bi L 能量分辨率为 ΔE 的三边光谱 /E :1 × 10 −4 测量 BFAx 室温下的 O 样品。为了获得最佳 XAFS 数据,BFAx O 粉在玛瑙研钵中研磨,然后与 BN 混合,最后压成颗粒。背景校正、归一化以及吸收光谱的前边缘和后边缘区域由 IFEFFIT 程序 [33] 内的 XAFS 数据处理软件 ATHENA 拟合。 E o 值由边缘区域中一阶导数的最大值确定。我们提取了 χ (k ) k 中的配置文件 0-12 Å −1 的空间 . k 3 × χ (k ) 配置文件被傅立叶变换为 R 0–8 Å 的空间,通过使用汉宁窗函数。 Fe2O3 和 Bi2O3 作为参考化合物进行测量。使用紫外-可见分光光度计(UV-Vis,UV 3900H)评估粉末的光学性质。在这项工作中,研究仅限于 0 ≤ x 的低掺杂浓度 ≤ 0.1。
结果与讨论
未掺杂 BFO (x =0) 和 BFAx O 粉末,从 2θ 扫描 15-60°的值,如图1所示。图1a中XRD图谱的全光谱表明所有样品都可以识别为相应菱形畸变钙钛矿结构的标准衍射数据(JCPDS Card File No. 20 -0169,空间群:R3c )。还可以看出,所有样品都显示出具有少量二次相的纯衍射图案。 x 在 27.6° 和 32.8°(Bi2Fe4O9 标记为“*”,Bi25FeO40 标记为“#”)处可以看到第二相的痕迹 =0.5 和 x =0.1 个样品,这可能是由于 Bi 在高烧结温度下的挥发性质 [34]。这在通过不同途径合成的 BFO 粉末中经常观察到 [35,36,37]。发现第二相在高掺杂值下连续增加。因此,B位Al替代Fe不能促进BFO的纯相;但是,样品的电性能不会受到影响。从图 1a 可以看出,掺杂样品的所有衍射峰首先移动到更高的 2θ 值与 Al 掺杂。为清楚起见,部分 2θ 中的 XRD 图 21 到 24° 的区域在图 1b 中被放大。从衍射峰的放大图可以看出,(101)衍射峰有明显的向高2θ移动 未掺杂 BFO 的值,这证实了 Al 成功掺杂到 BFO 的 B 位。与未掺杂的 BFO 类似,掺杂样品的 (101) 衍射峰经历了更高的 2θ 位移 首先是值,然后是较低的 2θ x 时的值 =0.1(被虚线包围),如图1c所示。 37-40°、50-52°和55-57°附近的放大XRD图如图1d所示。 XRD谱中有一些双峰,即(003)和(021),(113)和(211),(104)和(122)。随着Al含量的增加,这些峰的强度在x时先增大后减小 达到 0.1。众所周知,峰的强度通常与结晶度有关。衍射峰的减小表明 BFAx 的结晶度 O 减少。由于Al 3+ (0.51 Å) 的离子半径比 Fe 3+ 小 (0.65 Å),当掺杂量较小时,它很容易结合到 BFO 晶格中,但过多的掺杂离子会使 BFO 晶格不稳定。降低的结晶度可能是由于 Al 有利于产生更多的成核位点,这反过来又抑制了晶粒的生长。在其他掺铝 BFO 中也发现结晶度降低 [28, 30]。另一方面,如果有氧空位的产生和一些 Fe 3+ 的转化,就会发生这种情况。 到 Fe 2+ 由于 Al 3+ 在系统中造成的电荷不平衡 替代。在掺 Sr 的 BFO [38] 中也观察到了类似的现象。衍射峰的移动可能归因于晶胞收缩,因为与 Fe 3+ 相比,Al 的离子半径较小 . XRD结果表明,三价Al 3+ BFO中的取代不会导致可观察到的结构转变。
<图片>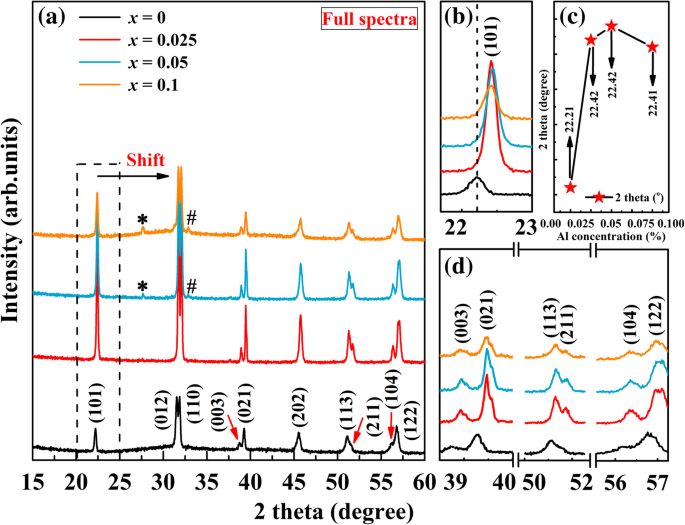
一 BFAx的XRD图谱 O (0 ≤ x ≤ 0.1)。 b XRD 图在 21-24° 范围内的放大视图。 c (101) 峰位置作为 Al 浓度的函数。 d 37-40°、50-52°和55-57°附近放大的XRD图
XRD 确认的结构也可以通过拉曼活性模式的位置和强度来表征。拉曼光谱对原子位移和分布很敏感。未掺杂的 BFO 和 BFAx 的拉曼散射光谱 图 2 给出了 O 粉末。根据群论,13 个光声子拉曼活动模式和 5 个非活动模式(即 5A 2) 预测了空间群为R3c的菱形畸变钙钛矿的BFO [39]。钙钛矿R3c晶格振动的理论分析 结构如下:
$$ {\varGamma}_{R3c}=4{A}_1\left(z,\kern0.5em {x}^2,{y}^2,\kern0.5em {z}^2\right)+ 5{A}_2\left(-\right)+9E\left(x,\kern0.5em y,\kern0.5em {x}^2-{y}^2,\kern0.5em xy,\kern0. 5em xz,\kern0.5em y\right) $$ (1)
一 BFAx的拉曼光谱 O (0 ≤ x ≤ 0.1)。 b 在 50–100 cm −1 范围内放大的拉曼光谱 和 c 125–200 cm −1
其中 A 是纵向光学 (LO) 模式和 E 是横向光模 (TO)。在图 2a 所示的全光谱中,在 RT 处清晰可见的拉曼活性模式的数量比预测的要少得多。在这项工作中,我们观察到了六种拉曼有源模式 (3A 1(LO) + 3E (TO)) 为 BFAx 哦粉。这可能是由于光谱中波段之间的意外简并以及无法从背景噪声中区分弱波段 [40] 或样品中的介电泄漏。对于未掺杂的 BFO,在 68.7 cm −1 处出现强而宽的峰 , 127.3 cm −1 , 和 164.9 cm −1 被分配给 A 1-1(LO), A 1-2(LO) 和 A 1-3(LO) 模式,分别。峰值在 204.7 cm −1 , 220.2 cm −1 , 和 251.8 cm −1 被分配给 E -1(TO), E -2(TO) 和 E -3(TO) 模式,分别(见图 2 中的表 1)。从光谱中可以清楚地看出这些E (TO) 模式是不可见的。众所周知,A 1-1(LO) 模式归因于 Bi-O 键,而倾斜 A 1-3(LO) 模式被视为 FeO6 八面体。 E (TO) 模式分配给 Fe-O 振动 [41]。拉曼结果证实所制备的未掺杂BFO属于空间群R3c的菱形畸变钙钛矿结构 .值得注意的是,所有四个样品都显示出相似的拉曼图案和振动模式。这表明相同的菱面体 R3c 空间群,但它们的强度和频率有些不同。 BFAx的拉曼图样全貌 O样本,A的峰值位置 1-1(LO) 模式略微偏移到更高的频率和 A 的峰值 1-3(LO) 模式变宽,表明掺杂剂 Al 将进入 BFO 的 B 位。 E 的强度 -1(TO), E -2(TO) 和 E 发现掺杂样品的 -3(TO) 模式略有增加。为清楚起见,A 1-1(LO) 和 A 50-100 cm −1 范围内的1-3(LO)模式 和 125–200 cm −1 如图2b和c所示。从这个放大的光谱中,很明显 A 随着铝含量的增加,1-1(LO) 模式表现出向更高频率的小偏移。与未掺杂的 BFO 相比,A 的峰值 掺杂样品的 1-3(LO) 模式被加宽。 A 的小偏移 1-1(LO) 模式,A 略微加宽 1-3(LO) 模式,以及一些 E 的强度变化 (TO) 模式可能与 Bi-O 和 Fe-O 共价键的变化以及 Al 掺杂样品中的压应力有关 [42, 36]。另一方面,上述所有变化可能是由于 B 位 Fe 3+ 离子已被 Al 3+ 部分取代 离子。显然,这些拉曼结果与 XRD 观察结果一致。 Goldschmidt 容差因子 (t ) 被广泛用于评估晶体结构的几何稳定性和畸变 [43],其中 t 由三种离子半径的比值定义,如下:
$$ t=\frac{\left({r}_{\mathrm{A}}+{r}_{\mathrm{O}}\right)}{\sqrt{2}\ \left({r} _{\mathrm{B}}+{r}_{\mathrm{O}}\right)} $$ (2)其中 r A是Bi 3+ 的半径 , r B是Fe 3+ 的平均半径 和 Al 和 r O是O 2- 的半径 .然而,Al 和 Fe 3+ 的半径 分别为 0.51 Å 和 0.65 Å,而 Bi 3+ 和 O 2- 半径分别为 1.03 Å(根据参考文献 [44])和 1.38 Å。 t 我们研究的钙钛矿复合材料 BFAx 的值 对于 x,发现 O 为 0.839、1.001、1.001 和 1.003 =0、0.025、0.05 和 0.1,分别(见图 2 中的表 1)。当 t 的值时,理想的 ABO3 化合物采用立方密堆积结构 是 1,而当 t <1 或> 1,产生几何应变 [45, 46]。随着Al浓度的增加,平均B位离子半径减小,导致t进一步增加 值从 0.839 到 1.003。这可能是由于 BFO 的低对称状态的微小变化。众所周知,钙钛矿中的B位阳离子被六个氧阴离子包围,当被更小的离子取代时,配位距离会减小。因此,为了清楚地分析铝取代的影响,有必要研究 BFAx 的局部电子结构 O 样品。
XAFS分为两种类型,即X射线吸收近边结构(XANES)和扩展X射线吸收精细结构(EXAFS)。在这个测量中,当 X 射线以距离 x 穿透板坯时 ,X射线束的强度将降低到I =我 oe - μx . XAFS 测量 X 射线的吸收作为 X 射线能量的函数 E ,即 X 射线吸收系数 μ(E ) =− d ln 我 /dx 由 X 射线束强度 I 的衰减确定 距离 x [47]。 I的比例 /我 o 绘制为 E 的函数 高于阈值 Fe K -edge (7112 eV) 和 Bi L 3 (13,419 eV),它可以提供关于 μ(E )。电子跃迁应遵循偶极选择规则。 X 射线吸光度 μ(ω ) 可由费米黄金法则得到,如下:
$$ \upmu \left(\omega \right)\propto \sum \limits_f{\left|\left\langle f\left|D\right|\left.i\right\rangle \right.\right|}^ 2\ \delta \left\langle {E}_i-{E}_f+\omega \right\rangle $$ (3)其中 |i ⟩ 是初始状态,|f ⟩ 是最终状态,D 是偶极算子,E 我 是 |i 的能量 ⟩, E f 是 |f 的能量 ⟩, 和 ω 是光子频率。 XANES 特征包含与吸收体的电子结构和原子结构的局部环境相关的有用信息。 XANES 函数 χ (E ) 定义如下:
$$ \chi (E)=\kern0.5em \left[\frac{\mu (E)-{\mu}_{\mathrm{o}}(E)}{\varDelta {\upmu}_{\ mathrm{o}}}\right] $$ (4)其中 μo(E ) 是平滑的类原子背景,Δμo 是归一化因子,由总原子背景吸收边的净增加引起。标准方程。 (4) 用于 XANES 数据处理过程中的背景减除和去除。首先,减去平滑的前边缘函数以从仪器中去除背景。其次,μ(E ) 被归一化,然后从 μ(E ) 得到 μo(E )。图 3a 显示了 Fe K 的全光谱 -未掺杂的 BFO 和 BFAx 的边缘 XANES 在这项工作中研究了 O 粉末和参考化合物 Fe2O3,而图 3b 报告了能量范围为 7100-7180 eV 的 XANES 光谱。从图 3b 可以看出,所有光谱的形状和峰值位置彼此相似。随着铝浓度的增加,吸收边缘略微向较低能量移动,这可归因于化学位移效应。随着电荷状态的降低,吸收边缘能量转移到较低的能量,表明 B 位的复杂键构型。众所周知,边缘偏移可用于获得平均氧化态。我们研究的样品 BFAx 的吸收边能 对于 x,发现 O 为 7124.95 eV、7123.87 eV、7123.84 eV 和 7123.80 eV =0、0.025、0.05 和 0.1,分别。对于参考化合物 Fe2O3,吸收边能量为 7126.14 eV(见图 3 中的表 2)。掺杂样品的吸收边低于 Fe2O3 (Fe 3+ ) 参考化合物。增加 x 值导致吸收边缘逐渐向 FeO (Fe 2+ ) [48]。由此可知,BFAx O 是混合价 (Fe 3+ /Fe 2+ ) 系统。另一方面,在所有四个样品中都观察到三个主要特征,前边缘峰A1和后边缘峰A2和A3。 BFO 的典型前缘峰 A1 对应于来自 O 1s 的电四极禁止跃迁 达到 Fe 3d 的,带有少量的 Al 3d 状态。随着 Al 浓度的增加,掺杂样品的前边缘峰 A1 的强度显示出小幅增加(见图 3c)。后边缘峰 A2 归因于 O 2p 带转移到 Fe 3d 轨道,所谓的配体到金属的电荷转移过程 [49],而峰 A3 是由 1s 到 4p 偶极子允许跃迁 [50]。随着Al浓度的增加,可以清楚地看到掺杂样品的这两个后边缘峰的强度增加(见图3d)。所有上述前缘和后缘峰强度的变化都可以从 Fe 3d 杂交之间的竞争来理解 和 Al 3d 与 O 2p 轨道。此外,Al离子掺杂后,有更多的未占位4p 发生在 BFAx 中的轨道 O.除此之外,整个光谱没有显示出明显的变化,这些结果证明Al离子部分掺杂到BFAx的B位 哦
<图片>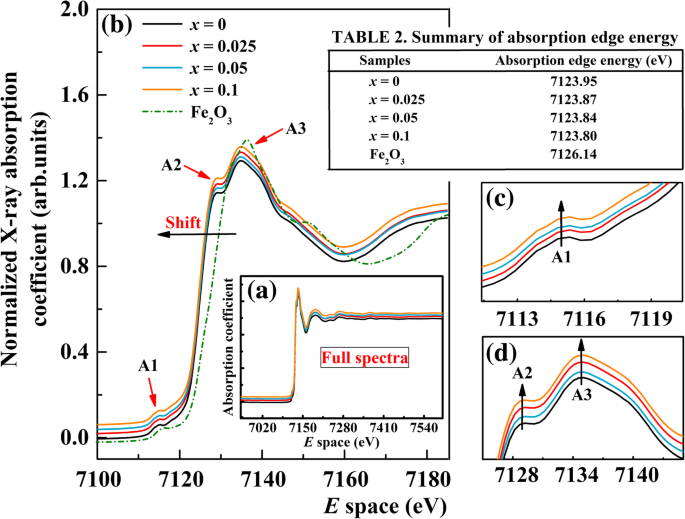
一 铁 K -BFAx 的边缘 XANES 光谱 O (0 ≤ x ≤ 0.1) 和参考 Fe2O3。 b XANES 光谱范围为 7100-7180 eV。 c 峰 A 的放大视图。d 峰B和峰C
通过拟合 k 获得 Fe-O、Fe-Fe/Al(即 Fe-O-Fe/Al)和 Bi-O 键分布 3 -加权 (k 3 × χ (k )) 原始数据,如下:
$$ k=\sqrt{\frac{2m\left(E-{E}_{\mathrm{o}}\right)}{\mathrm{\hbar}}} $$ (5)其中 E o 是吸收边能量,ħ 是普朗克常数。使用标准 EXAFS 方程进行进一步拟合(一些近似和振荡),如下所示:
$$ \chi (k)=\sum \limits_R{S}_{\mathrm{o}}^2{N}_{\mathrm{R}}\frac{\left|f(k)\right|} {k{R}^2}\sin \left(2 kR+2{\delta}_{\mathrm{c}}+\phi \right){e}^{\frac{-2R}{\lambda ( k)}}{e}^{-2{\sigma}^2{k}^2} $$ (6)其中 \( {S}_{\mathrm{o}}^2 \) (\( 0<{S}_{\mathrm{o}}^2<1 \)) 是缩减因子,N R 是距离 R 处的背向散射原子数 , f (k ) 是后向散射幅度,δ c 是中心的相移,ϕ 是背向散射原子,λ 是核孔寿命,σ 2 是来自多个距离的 Debye-Waller 因子,并且 k 是光电子平均自由程。 EXAFS 区域通常是指边缘跳跃上方 20-30 eV 的能量范围,它对材料中的短程有序类型、键距和配位数很敏感。虽然,方程。 (6) 可以提供有关 EXAFS 近似值和振荡的一些信息,但它不是一种用于可视化 EXAFS 频谱信息内容的特别方便的形式。因此,傅里叶变换可以用来分解一个k 空间信号转换成其不同的组成频率 [32]。傅里叶变换是原子间距离R的复函数 ,其幅度由χ的实函数表示 (R )。在此函数中,峰的位置与键距和相邻离子有关。方程(6)可以从 k 转化而来 R 的空间 空间由傅立叶变换,如下:
$$ \chi (R)=\frac{1}{\sqrt{2\pi }}{\int}_{k_{\mathrm{min}}}^{k_{\mathrm{max}}}\omega (k){k}^n\chi {e}^{-2 ikR} dk $$ (7) $$ \left\{\begin{array}{c}\genfrac{}{}{0pt}{} {\kern2.5em 0\kern7.5em k<{k}_{\mathrm{min}}\ }{\sin^2\left[\frac{\pi \left(k-{k}_{\mathrm {min}}\right)}{2\left({k}_2-{k}_{\mathrm{min}}\right)}\right]\kern3.25em {k}_{\mathrm{min} }其中 k 最大值和 k min 是变换后的 k 的最大值和最小值 空间,分别为 χ (R) 是汉宁窗函数,ω (k ) 是高斯窗函数,k n 是权重因子 (n =0, 1, 2, 3)。 k 2 和 k BFAx 的 3 个值 O分别为2和10,n的值 是 2. Fe K 的傅里叶变换 -BFAx 的边缘 EXAFS 执行 O 个样本,如图 4a 所示。由于氧阴离子的散射,以~ 1.503 Å 为中心的不对称峰(由第一条虚线包围)被确定为 Fe-O 键。 ~ 3.527 Å 附近的第二个强峰(由第二条虚线包围)对应于 Fe-Fe/Al 键,这可以通过氧阴离子从紧邻的 Fe/Al 原子的散射来解释贝壳。图 4 中的表 3 总结了第一和第二配位壳的距离。从峰值位置来看,与未掺杂的 BFO 样品相比,Fe-O 和 Fe-Fe/Al 键倾向于略微向更小的 移动 随着 x 增加的值 .这表明Al离子的掺杂不仅影响中心Fe原子的最近邻局域结构,而且影响Fe原子的次近配位壳层。另一方面,Fe-O 键转移到更小的 R 值可能是由于 Al 离子的半径小于 Fe 离子的半径。这与XRD数据一致。 Fe-Fe/Al 向更小的 R 移动 值还表明平均 Fe-Fe/Al 键长(其中两个 Fe 3+ 离子位于相邻氧八面体的中心)逐渐变短,并且在掺杂铝的样品中键角发生变化。然而,掺杂样品的 Fe-O 分布的峰值强度略有增加,而 Fe-Fe/Al 分布的峰值强度几乎没有变化。这表明 Fe-O 的铁邻体结构已被 Al 掺杂改性。 Al掺杂样品中较短的Fe-Fe/Al键可以解释为什么XRD中的主要峰(101)向更高的2θ移动 角度(见图 1b)。这表明 Al 取代 Fe 会影响氧八面体,这进一步降低了两个相邻 Fe 原子之间的配位距离。图 4b 显示了 Fe K -BFAx 的边缘 EXAFS 在 k 上处理的 O 个样本 3 × χ (k ) 以 k 振荡 0-10 Å −1 的空间 .可以看出,所有的 k 3 × χ (k ) spectra show similar patterns at the smaller k values but different at larger k values with some various noise (surrounded by the dash line). Al-doped samples show a broader k 2 × χ (k ) spectrum than those of undoped BFO in the k space of 8.2–9.3 Å −1 , implying an enhanced short-range structural disorder in BFAx O samples. The noises are observed in a k space of 10–10.4 Å −1 . These changes indicated that the local structure of center atoms has changed due to B-site Al doping, similar to what was reported by Li et al. [51].

一 Fourier transforms of Fe K -edge k 3 -weighted EXAFS data, for the BFAx O (0 ≤ x ≤ 0.1). b EXAFS χ (k ) × k 3 spectra of BFAx O (0 ≤ x ≤ 0.1). The spectra were aligned along the Y -axis for better comparison
The peak positions, intensities, and shapes of the line in the Bi L 3-edge XANES spectrum are well known to depend on the local electronic structure of the Bi atoms, which could provide information on the Bi valence. The full spectra of Bi L 3-edge XANES of undoped BFO and BFAx O samples are also shown in Fig. 5a, while Fig. 5b shows the Bi L 3-edge XANES spectrum in the energy region of 13,400–13,480 eV. The analysis of these spectra helps to investigate the local electronic structure of Bi ions in the doped system. From Fig. 5b, it can be seen that the shape of all spectra are the same to each other and there is almost no change of absorption edge in the whole series. The absorption edge energies for our study are found to be 13,429.1 eV, 13,429.4 eV, 13,429.3 eV, 13,429.3 eV, and 13,429.8 eV for x =0, 0.025, 0.05, and 0.1 and the reference compound Bi2O3, respectively (see Table 4 in Fig. 5). The absorption edges slightly shift toward higher energies with increasing Al concentration. The absorption edge in the Bi L 3-edge of the BFAx O samples matches well with that of the reference compound Bi2O3, which indicates that the valence state of Bi ions in all the samples is in + 3 valence state. However, there are two post-edge peaks found in all samples and marked as B1 and B2, respectively. These two post-edge peaks are caused by the electric-forbidden transition from 2p 3/2 level to the 6d 那些。 Compared with undoped BFO, the intensity of peak B2 can be clearly seen to increase for the doped samples (see Fig. 5c), which means the transition from 2p 3/2 to 6d state increases, so does the energy of 6d state. Except these, there is no other significant change in the whole spectrum.

一 Bi L 3-edge XANES spectra of BFAx O (0 ≤ x ≤ 0.1) and reference Bi2O3. b XANES spectrum in the range of 13,400–13,480 eV. c Enlarged view of the peak D and peak E
Fourier transform of Bi L 3-edge EXAFS radial distribution functions is performed, as shown in Fig. 6a. A high-intensity peak located at around 1.618 Å corresponds to the nearest Bi-O coordination shell (surrounded by the dash line), which is a result from scattering from the nearest-neighbor atomic shell of Bi, i.e., oxygen anions. However, the position of Bi-O bond shifts toward larger R values for the doped samples (see Table 5 in Fig. 6). This indicates that the substitution of Al for Fe could affect the nearest-neighbor local structure of the central Bi atom. It also indicates the extension of Bi-O bond length. The peak intensity of the Bi-O distribution exhibited a small increase with increasing Al content, which suggests that the iron-neighboring structure of Bi-O has changed. Figure 6b shows the Bi L 3-edge k 3 × χ (k ) EXAFS spectra with a k space of 0–14 Å −1 . From Fig. 6b, it can be seen that all the spectra shape shows similar patterns except some error noises. The error noises are observed in the k space of 12–14 Å −1 (surrounded by the dash line). This result may imply that the k 3 × χ (k ) EXAFS function of the center Bi atoms has changed with Al doping. This also suggests that the B-site Al substitution influences short-range structural disordering.

一 Fourier transforms of Bi L 3 -edge k 3 -weighted EXAFS data, for the BFAx O (0 ≤ x ≤ 0.1). b EXAFS χ (k ) × k 3 spectra of BFAx O (0 ≤ x ≤ 0.1). The spectra were aligned along the Y -axis
The optical properties of the samples are studied by using RT UV-Vis, which is used to characterize the optical properties of the materials. The UV-Vis absorption spectra of undoped BFO and BFAx O samples in the wavelength of 300–800 nm are shown in Fig. 7a. As a result, the UV-Vis spectra of the undoped BFO and BFAx O samples show two absorption edges (marked by dashed arrows). One is a band around 650 nm, which is due to the metal-to-metal transition. The other is a band around 760 nm, which is caused by crystal field transition [52]. In addition, the strong absorption band is observed at about 490 nm (marked by dashed arrow), which is attributed to the electronic transition from O 2p to Fe 3d state in the BFO. These strong bands indicate that the BFO prepared by hydrothermal method could be a promising visible-light photocatalytic material. BFO is of direct transition with a value of n as 2. The absorption edge of the doped samples shifted from 659 to 619 nm, suggesting that the BFAx O powders absorb visible light in the wavelength range of 600–659 nm (see Fig. 7a). A similar blue-shift phenomenon was observed earlier in other element-doped BFO [53,54,55]. This blue shift in the absorption spectra of Al-doped samples in comparison with the undoped BFO shows that doping Al causes a change in the local structure for BFO. From Fig. 7a, one can see that the absorption spectra of the Al-doped samples exhibit a sharp increase around 490 nm and it suggests that all samples can absorb remarkable amounts of visible light. For the sample with x =0.025, the absorption spectrum shows a sudden increase. It means that it has a wider absorption range than the other samples in this range of visible light. The optical band gap of the samples has been calculated by Tauc’s formula, as follows:
$$ ahm=A{\left( hm-E\mathrm{g}\right)}^n $$ (9)
一 UV-Vis absorption spectrum of BFAx O (0 ≤ x ≤ 0.1). b Plots of (ahν ) 2 vs. photon energy. c E g values as a function of Al concentration
where a is the absorption coefficient, A is the parameter, h is the Planck’s constant, m is the frequency of the incident photon, E g is the optical band gap, and n (for direct n =2, for indirect n =0.5) is a constant associated with different types of electronic transitions, as shown in Fig. 7b. The calculated E g values are found to be 1.833 eV, 1.888 eV, 1.866 eV, and 1.905 eV for x =0, x =0.025, x =0.05, and x =0.1 samples, respectively. It is easy to see that the band gap increases with the substitution ratio, as shown in Fig. 7c. The increase in the band gap is attributed to the doping effect. The E g value for the undoped BFO is about 1.833 eV, which is lower than the previous reports [56, 57].
Conclusion
In summary, the BFAx O (x =0, 0.025, 0.05, and 0.1) multiferroic powder samples were successfully synthesized via hydrothermal route. Effects of Al substitution on the structural, electrical, and optical properties of the samples were studied. The structural study reveals that Al-doped BiFeO3 shows the existence of secondary phases and lattice contraction due to lower ionic radii of Al doped into B-site, which still retains its rhombohedral R3c perovskite structure. Raman scattering measurement infers six Raman active phonon modes, which further confirms the result of XRD. XAFS studies on the Fe K -edge and B L 3-edge of the BFAx O samples and of the reference compounds Fe2O3 and Bi2O3 were performed, and the obtained results were compared in order to determine the valance states of Fe and Bi ions in the system. The Fe K -edge XAFS results revealed that BFAx O is a mixed-valent (Fe 3+ /Fe 2+ ) system. The results of Fe K -edge XAFS also illustrate a competition between the Fe 3d and Al 3d orbitals on hybridization with the O 2p and occurrence of the more 4p orbitals with Al doping. Besides, Al ion doping affects both the nearest-neighbor and next-nearest coordination shells of the Fe atom. The B L 3-edge XAFS results indicate that valence states of Bi ions in all the samples are in + 3 and the transition from 2p 3/2 to 6d state and the energy of 6d state increases. Substitution Al for Fe could affect the nearest-neighbor local structure of central Bi atom. The BFAx O prepared by hydrothermal method could be an appropriate visible-light photocatalytic material due to a strong absorption band in the visible region.
缩写
- AFM:
-
Antiferromagnetic
- BFAx O:
-
BiFe1-x Alx O3
- BFO:
-
BiFeO3
- EXAFS:
-
X-ray absorption fine structure
- FE:
-
Ferroelectric
- FM:
-
Ferromagnetic
- RT:
-
室温
- 紫外可见光:
-
Ultraviolet-visible
- XAFS:
-
X-ray absorption fine structure
- XANES:
-
X-ray absorption near edge structure
- XRD:
-
X射线衍射
纳米材料
- (p-i-n) 结 GaAs 纳米线太阳能电池中的等离子体增强光吸收:FDTD 模拟方法研究
- X 射线同步加速器粉末衍射探测到的 HoCo0.5Cr0.5O3 异常热膨胀
- 新型纳米粒子增强蠕虫状胶束系统的研究
- 通过角分辨 X 射线光电子能谱研究 Al2O3 封端的 GaN/AlGaN/GaN 异质结构的表面极化
- 硫酸软骨素-甲氨蝶呤纳米凝胶的抗肿瘤研究
- 螺旋型天线微桥结构太赫兹微测辐射热计的调频和吸收改进
- InSe 纳米带的电子结构和 I-V 特性
- Al-Doped ZnO 薄膜在红外区域的光学特性及其吸收应用
- GaAs/AlAs 超晶格点缺陷的第一性原理研究:相稳定性以及对能带结构和载流子迁移率的影响
- 过渡金属在黑磷烯上的吸附:第一性原理研究
- Ti3C2Tx MXene 的原位高压 X 射线衍射和拉曼光谱研究
- 用于臭氧检测的腔增强吸收光谱 (CEAS)