

高效独立激发蓝色发光碳点
摘要
蓝色发光碳点(CDs)是通过水热法合成的。当 CD 溶液的浓度降低时,观察到最大发射波长从 480 nm 蓝移到 443 nm。低浓度 CDs 的光致发光 (PL) 光谱显示出与激发无关的行为,这与之前的报道大不相同。两种不同的发光机制可能起作用:来自 sp 2 的本征发光 -碳网络可能是低浓度下发射的较短波长部分(与激发无关)的原因,纳米簇的高极性导致较长波长部分在高浓度 CD 溶液中的激发相关行为。 CDs的光物理性质和浓度依赖性行为从实验和机制的角度为CDs提供了新的见解,这将在不久的将来促进CDs的多种潜在应用。
背景
碳点作为碳纳米材料家族中的一种荧光材料,在过去几年中引起了越来越多的关注。通常,CD具有石墨或无定形碳骨架的核心,其表面涂覆有含氧基团、聚合物和其他物质[1]。同时,不大于 10 nm 的 CDs 具有独特的光物理特性,如高光稳定性、良好的生物相容性、优异的光学性能和低环境危害 [2, 3]。受这些特性的启发,CDs 具有各种潜在的应用,如药物输送 [4]、荧光墨水 [5]、传感器 [6, 7]、光电子学 [8]、光催化 [9, 10] 和发光器件[5, 11,12,13]。迄今为止,已经开发了多种合成方法来制备CDs,如石墨电化学氧化[9]、水热法[5, 10]和微波辅助合成[14, 15]。
CD 的一项特殊性质是发射峰与激发波长的相关性。在不同的激发波长下,CD 具有从紫色到红色的不同光致发光 (PL) 峰 [10]。已经报道了许多可能的原因来解释这种现象,例如尺寸 [9, 11]、元素掺杂 [10, 14]、溶剂极性 [16]、缺陷、表面状态 [17]、表面基团 [18, 19] 或表面钝化 [20]。然而,很少观察到CDs的激发独立性。
有趣的是,我们发现通过用去离子水稀释 CD 溶液,观察到最大发射峰从 480 到 440 nm 的蓝移。此外,CDs 的发射强度随着浓度的降低而变得更强。随着激发波长的变化,PL 光谱在 443 nm 处显示出一个不变的发射峰,这与之前的报告非常不同。纳米簇的高极性与sp 2 -碳网络可能是造成这些现象的原因。
方法
试剂和化学品
SCR(中国上海)需要一水柠檬酸(99.5%),而乙二胺则来自天正试剂(中国天津)。去离子水来自电阻率为 18.25 m Ω cm 的净水器净水系统(中国四川)。所有化学品均按原样使用,无需进一步纯化或处理。
碳点的制备
如下制备 CD:将柠檬酸 (1.0507 g) 和乙二胺 (335 μL) 添加到去离子水 (10 mL) 中。然后,将充分搅拌的溶液转移到衬有聚四氟乙烯的高压釜中。将溶液加热至 150°C 并保持 5 小时。反应结束后,将反应器自然冷却至室温。制备的CD溶液颜色为淡黄色。在表征之前,CD 溶液通过以下方法处理:取 1 ml 原始 CD 溶液,然后用 5-400 ml 去离子水稀释。 CD溶液稀释后颜色由黄色变为无色。
特征化
使用 Hitachi F4500 荧光分光光度计和具有 325 nm He-Cd 激光器的共焦拉曼显微镜进行光致发光。吸收光谱由 Shi-madzu UV-3101PC 光谱仪收集。傅里叶变换红外 (FTIR) 用 Brucker VERTEX 光谱仪记录;透射电子显微镜 (TEM) 图像记录在 FEI Tecnai G2 20S-twin 上。使用 Malvern Zetasizer Nano ZS 进行动态光散射 (DLS) 研究。使用 Bruker D8 系统收集 X 射线衍射 (XRD) 图案。使用爱丁堡 FLS920 荧光光谱仪研究荧光衰减曲线。拉曼光谱在 LabRAM HR Evolution (Horiba) 上进行,激光激发波长为 532 nm。 X 射线光电子能谱 (XPS) 分析由 PHI 5000 Versa Probe(ULVAC-PHI,日本)测量。采用多模式扫描探针显微镜(MM-SPM)进行原子力显微镜(AFM)测量。
结果与讨论
通过透射电子显微镜、X 射线衍射 (XRD) 和拉曼光谱测量证实了 CD 的形成。如图 1a 所示,获得平均直径约为 3.6 nm 的球形碳纳米颗粒。插图显示了 2.5 和 5 纳米之间的粒径分布。图 1b 显示 CD 具有晶格间距为 0.295 nm 的结晶内核,这对应于石墨碳的 (002) 平面 [4, 9, 14]。 TEM 图像中可辨认的 CD 晶格结构表明所得纳米颗粒具有石墨内核。 CD 的 XRD 衍射图显示在 20.24° 处有一个宽峰(附加文件 1:图 S1),接近石墨结构的 (002) 层间距 [5, 21]。 1598 cm −1 处的 G 带 和 D 波段在 1350 cm −1 CDs 在拉曼光谱上并不明显(附加文件 1:图 S2)。 CD 的强荧光可能会干扰拉曼表征。此外,两个峰的缺失进一步证明了CDs由纳米晶类石墨核和无序sp 3 组成。 -碳[21]。
<图片>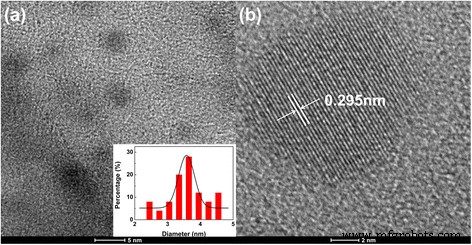
制备的 CD 的 TEM 和 HRTEM 图像。 一 制备的 CD 的透射电子显微镜 (TEM) 图像(插图 显示粒度分布)。 b 一张代表性 CD 的高分辨率 TEM 图像,显示其结晶石墨内核
如图 2a 所示,制备的 CD 水溶液呈淡黄色(左),在 365 nm 紫外光(右)激发下呈现亮蓝色发光。在CD溶液的吸收光谱中,243 nm处的吸收峰归因于\( \pi \)→\( \pi \) * C=C,345nm处的吸收峰对应于n→\( \pi \) * C=O 键的转变(图 2a)[14]。 CD 的傅里叶变换红外光谱 (FTIR) 光谱(图 2b)表明其表面存在丰富的含氧基团。如图 2b 所示,1120 cm -1 处的峰值 可以归因于 C-O-C 的不对称和对称伸缩振动。 1445 和 1464 cm −1 处的峰值 分配给 C-H 弯曲振动; 1488 cm −1 处的峰值 表明存在N-H弯曲振动; 1689 cm −1 处的峰值 归因于C=O伸缩振动; 2935 cm −1 处的峰值 由甲基/亚甲基的 C-H 伸缩振动引起;宽频带以 3100–3500 cm −1 为中心 分配给 O-H 和 N-H 伸缩振动 [5, 10, 14]。 FTIR 分析结果证实所制备的 CDs 表面存在含氧基团,例如 C =O 和 -OH。 XPS 调查进一步支持了 FTIR 分析。如附加文件 1:图 S3 所示,CD 主要由碳、氧和氮元素组成。 C 1s 的高分辨率 XPS 光谱在 284.56、285.66 和 287.7 eV 处显示三个峰,表明存在 C=C/C-C、C-O 和 C=O。 N1s 的高分辨率光谱表明存在类吡咯 N (399.7 eV) 和类石墨/氨基 N (400.7 eV)。 O 1s 高分辨率光谱在 531.55 和 532.31 eV 的两个峰归因于 C=O 和 C-OH/C-O-C 键 [21,22,23,24]。 XPS 分析的结果与 FTIR 光谱非常吻合。结合所有这些表征数据,认为CDs由纳米级石墨状核和核表面的含氧基团组成。
<图片>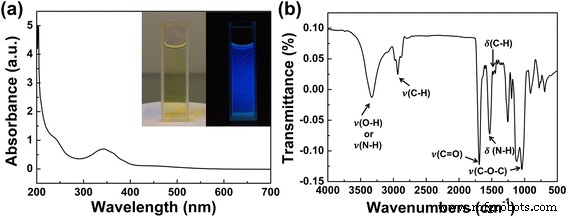
制备的 CD 的 UV-vis 吸收光谱和 FTIR 光谱。 一 CD的紫外可见吸收光谱。 插图 照片显示了在自然光下准备好的 CD(左 ) 和 365 nm 照射下(右 )。 b CDs的FTIR光谱
用 5 ml 去离子水稀释的 CD 的发射光谱显示出典型的激发相关特征。随着激发波长逐渐增加,PL 峰向更长的波长移动(图 3a;激发波长从 330 到 480 nm 逐渐增加,330-390 nm 的发射强度乘以 25),这与其他报道一致[1, 5, 14]。 CD 在 481 nm 处具有最大发射强度,激发波长为 420 nm。使用硫酸氢奎宁(0.1 M H2SO4 中的 QY 0.56)作为参考,CD 的量子产率为 74.8%。高量子产率应该是 CDs 的分子态 [5]。此外,所获得的 CD 的发射波长和 PL 强度都对加水量敏感;换句话说,它们对 CD 溶液的浓度很敏感(图 3b-d)。这一结果与其他方法合成的 CDs 不同,后者仅显示发射峰随着 pH 值的变化而略有移动 [25]。通过将更多的去离子水(10、25、50、100、200、300 和 400 毫升)添加到 1 毫升制备的 CD 溶液中,观察到发射峰的蓝移从 480 到 440 纳米(附加文件 1 :图S4),而CD溶液的相应吸收光谱没有变化(附加文件1:图S5)。 330 至 400 nm 范围内的发射峰强度逐渐增强,而 420 至 480 nm 范围内的发射峰强度逐渐消失(附加文件 1:图 S4)。当具有不同浓度的 CD 被相同的 330 nm 波长激发时,可以在图 3b 的归一化 PL 光谱中清楚地看到这种蓝移。在图 3b、c 中,发射波长的变化主要发生在加入的去离子水的体积小于 25 毫升时,从 505 到 450 纳米不等。随着进一步稀释,发射波长的变化非常小。在图 3c 中,发射峰的 PL 强度随着 CD 浓度的降低而不断增加。这种强度增强可能得益于高浓度溶液中碰撞猝灭和自吸收猝灭的减少[5, 26]。
<图片>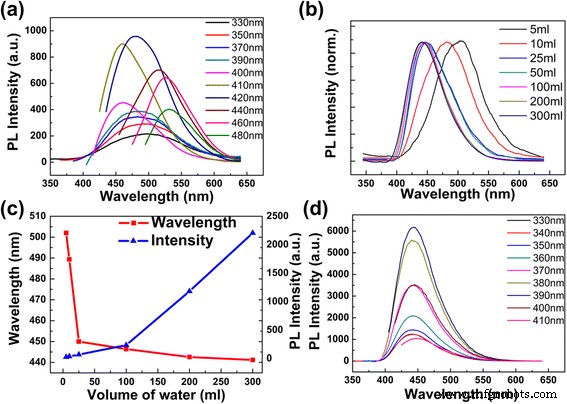
CDs在不同体积去离子水中的PL光谱。 一 1 ml 制备好的 CD 与 5 ml 去离子水(pH 10.41)的 PL 光谱。 b CDs 在不同体积水中的归一化荧光发射光谱,激发波长为 330 nm。 c 最大PL强度和发射峰作为不同加水体积的函数。 d 用 300 ml 去离子水稀释 1 ml 制备的 CDs 溶液的发射光谱
用 300 ml 去离子水稀释后,PL 光谱在 443 nm 处显示单发射峰,该峰随着激发波长的变化而不变(图 3d;激发波长从 330 到 410 nm 逐渐增加)。在 390 nm 激发波长下获得最高发射强度。即使用更多体积的水稀释(附加文件 1:图 S4),发射光谱也不会偏移。
当将不同体积的去离子水加入制备好的 CD 溶液中时,溶液的 pH 值会发生变化。我们观察到的现象可能是由不同的 pH 值引起的。为了验证pH值是否是造成该现象的主要原因,对不同pH值的CD溶液进行了详细分析。用 5 ml 去离子水稀释的 CD 溶液的 pH 值为 10.41。用 300 毫升去离子水稀释制备的 CD 溶液时,pH 值变为 10.2。然后,我们通过加入 NaOH 将 300 毫升稀释的 CD 溶液的 pH 值从 10.2 调整到 10.41。图 4a 显示了将 pH 值调整到 10.41(激发波长从 330 到 410 nm 逐渐增加)后 CD 溶液的 PL 光谱。从两幅图(图 3d 和 4a)中,我们可以明显地注意到,即使将 pH 值从 10.2 调整到 10.41,PL 峰的位置和强度几乎不变。然后,我们通过添加 NaOH 将含有 5 ml 去离子水的 CD 溶液和含有 300 ml 去离子水的溶液的 pH 值调整为相同的 12.08(在图 4b 中,激发波长从 330 到 480 nm 逐渐增加) ,330-380 nm 的发射强度乘以 15,390 nm 的发射强度乘以 6;在图 4c 中,激发波长从 330 到 410 nm 逐渐增加);与图 3a、d 相比,PL 发射的位置和强度也没有变化。上述结果表明,pH 值不是我们实验中发射峰不变的原因。由此可见,浓度是调节发射波长和固定发射峰的关键。据我们所知,这是第一个表明通过用去离子水调节CD溶液的浓度可以很容易地调节CD的发射波长和PL强度的报道。
<图片>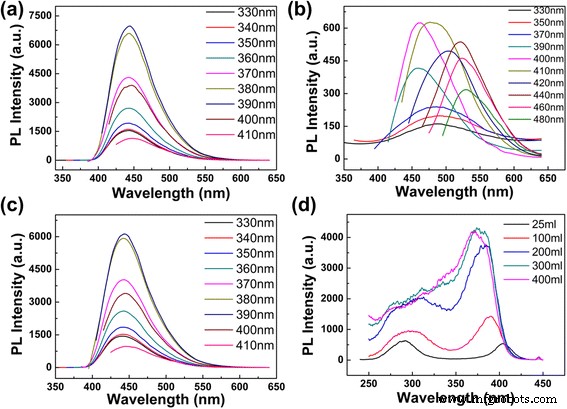
CDs在不同体积的水和pH值下的发射和激发光谱。 一 CD 与 300 毫升去离子水 (pH 10.41) 的发射光谱。 b CD 与 5 ml 去离子水 (pH 12.08) 的发射光谱。 c CD 与 300 毫升去离子水 (pH 12.08) 的发射光谱。 d CDs在不同体积水中的445 nm激发光谱
为了深入了解 CD 的激发独立特性,我们测量了不同发射波长的激发光谱,并在附加文件 1:图 S6 中显示。对于高浓度的 CD 溶液(用 25 ml 去离子水稀释 1 ml 制备的溶液),有两个强烈的激发峰分别位于 290 和 400 nm。随着浓度从高到低,290 nm 处的激发峰变弱,400 nm 处的峰增强,蓝移至 370 nm(图 4d)。不同浓度激发光谱的特征表明,CDs的发光可能有多个中心。为了进一步证明,使用 280 nm 的激发波长和 447 nm 的发射波长测量 CD(用 25 ml 去离子水稀释的 1 ml 制备溶液)的荧光寿命(附加文件 1:图 S7)。平均寿命为 11.85 ns,衰减曲线可以通过双指数函数拟合,寿命分别为 5.11 ns (35.08%) 和 13.28 ns (64.92%)。样品的多重寿命可能是由于样品表面存在不同的荧光团或能级[18]。
一些 CD 研究表明,即使使用稀释溶液,也存在小颗粒和颗粒聚集体 [27]。 Iijima [28] 也观察到类似类型的聚集,其中发现小碳颗粒聚集成 80 纳米大小的纳米角结构。小颗粒通过范德华力相互吸引。我们通过动态光散射 (DLS) 测量(附加文件 1:图 S8)估计了不同体积去离子水的 CDs 的粒径,结果表明 CDs 的流体动力学直径不同,范围从 34 到15 纳米。在制备好的 CDs 溶液(高浓度)中,平均直径为 34 nm。用 100 毫升去离子水稀释后,CD 的平均直径为 15 纳米。水溶液中 CD 的平均尺寸随着浓度的降低呈下降趋势(附加文件 1:图 S8)。因此,可以得出结论,在高浓度下,许多单个 CD 聚集在一起形成纳米级簇,导致平均直径增加。单个 CD 和纳米级簇在解决方案中共存。在低浓度下,纳米级簇已分离成单个 CD。通过 DLS 测量测试的 CD 的平均尺寸大于 TEM 结果(4-6 nm),这主要是因为 DLS 考虑了包括颗粒以及吸收的分子和离子的整体流体动力学直径 [27]。测量了不同体积水的 CD 的原子力显微镜 (AFM)。如附加文件 1:图 S9 所示,当浓度较高时,图像显示单个 CD 聚集在一起形成纳米级簇,平均直径为 40 nm;随着浓度从高到低,纳米团簇逐渐分离成单个CD,测得的直径约为10 nm,小于40 nm,与DLS结果一致。
在水介质中由有机材料形成 CDs 的共同观点是 CDs 由 sp 2 的纳米晶核组成 - 杂化二维石墨烯型岛 [10, 29] 被 sp 3 破坏 - 杂化钻石型内含物 [27, 29]。在纳米粒子的形成过程中,源自起始材料的极性基团附着在 CD 的表面,这使得粒子可溶于水。这一观点得到了从不同起始材料获得的 CD 的拉曼光谱 [27, 30] 的证实,这证明了 sp 2 的存在 - 和 sp 3 - 相似比例的混合结构。同时,所有研究的通过有机材料热处理获得的水溶性CDs都含有羟基、羧基和羰基形式的氧元素[16]。粒子表面的极性基团对CDs的发射尤为重要[16,18,31]。
从上面的实验中,我们可以得出结论,CD 溶液中有两种不同的发光物质。 sp
2
的本征发光 -碳网络和纳米级簇的高极性可能导致不同的发射现象(图 5)。由于其极性表面基团,例如 -CO 和 -OH [15, 18, 31](图 2b),发现单个 CD 的行为类似于电偶极子 [32]。 CD 表面上的含氧基团可以负责荧光发射的较长波长部分 [19]。当高浓度时,许多CDs通过范德华力聚集在一起[28]形成纳米团簇,然后大量-CO和-OH聚集在一起,导致纳米团簇表面的极性更高[15] ]。纳米级簇的高极性导致依赖于激发的特性 [15, 19, 31]。同时,纳米簇的高度氧化和更高的极性导致电子从激发态快速弛豫到亚态,这对应于更长的波长。然后,亚态有助于光发射,最终产生更长波长的发射 [15]。因此,在高浓度时,在较长波长部分会出现激发依赖现象。在配制好的CD溶液中加入去离子水后,溶液的浓度逐渐降低。然后,形成纳米团簇的CDs被分离并重新分散成单个CDs,导致极性减弱和更长波长的发射光谱消失。此外,簇的分离也会导致 290 nm 处的激发峰消失(图 4d)。与单个CD表面极性基团(-CO和-OH)的发射相比,sp
2
的本征发光 -碳网络随着CD浓度的降低起主导作用。低浓度时,单张CD的荧光光谱不对称,向高能区(短波长)展宽,如图4a所示[15, 33]。> <图片>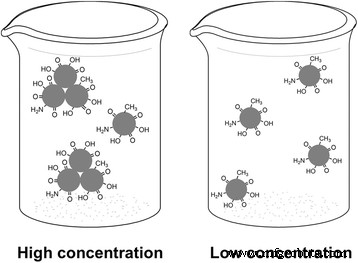
高浓度和低浓度 CD 的示意图。 左 :在高浓度时,许多单个CD形成纳米级簇; 对 :低浓度时,纳米团簇分离成单个CD
结论
总之,我们已经通过水热法合成了 CD。值得注意的是,所制备的 CD 具有优异的水分散性和独特的 PL 特性,例如浓度敏感性和与激发无关的发射波长。当 CD 的浓度从高到低时,观察到最大发射波长从 480 到 443 nm 的蓝移。低浓度 CD 的 PL 光谱显示出与激发无关的行为,发射峰位于 440 nm,这与之前的报告非常不同。可以得出结论,存在两种不同的发射机制。 sp 2 的本征发光 -碳网络负责低浓度下短波长(与激发无关)的发射,纳米簇的高极性导致高浓度下较长波长部分的激发依赖性。 CDs 良好的光物理特性和浓度依赖性行为将提供一种方法来调整发射波长,并从实验和机制的角度提供对 CDs 的新见解,这将在不久的将来促进 CDs 的多种潜在应用。
缩写
- 原子力显微镜:
-
原子力显微镜
- CD:
-
碳点
- DLS:
-
动态光散射
- FTIR:
-
傅里叶变换红外
- PL:
-
光致发光
- TEM:
-
透射电子显微镜
- XPS:
-
X射线光电子能谱
- XRD:
-
X射线衍射
纳米材料
- 琥珀色
- 碳 M2
- 制造和成像环碳
- 重吸收抑制的 II 型/I 型 ZnSe/CdS/ZnS 核/壳量子点的合成及其在免疫吸附测定中的应用
- 合成富含吡啶的 N、S 共掺杂碳量子点作为有效的酶模拟物
- 基于苯基三甲氧基硅烷改性氧化铝纳米颗粒的 Al2O3:SiOC 纳米复合材料的形成和发光特性
- 磁性碳微球作为可重复使用的吸附剂从水中去除磺胺
- 从豆腐废水中合成荧光碳量子点的简单方法
- 掺杂铕的核-壳 ZnSe/ZnSe 量子点中粒子间能量转移的壳厚度依赖性
- 利用柠檬汁通过水热反应制备的荧光碳量子点的材料和光学特性
- 用于光热疗法的聚多巴胺碳点的简便一锅法合成
- ATL Composites 为混凝土支柱修复项目做出贡献