

合成富含吡啶的 N、S 共掺杂碳量子点作为有效的酶模拟物
摘要
通过微波固相热解在 50 秒内合成了具有高 N 和 S 掺杂水平的 N 和 S 共掺杂碳量子点(N,S-CQD)。由于主要的吡啶氮注入共轭骨架,同时实现了高酶模拟催化活性和光致发光量子产率。
背景
作为一种新型零维碳材料出现的碳量子点 (CQD) 因其高化学稳定性、低细胞毒性以及独特的电子性质和光学行为而受到极大关注 [1,2,3]。由于具有–OH 和–CO2H 等活性表面基团,CQD 可以与其他有机物或无机物重新结合,用于各种奇妙的应用,包括生物成像 [4, 5]、光电器件和用于降解有机染料或从中产生氢气的光催化剂。水分解 [6,7,8]。最近,实验和理论结果均证实杂原子掺杂是改善 CQD 电子和光学性能的有效方法 [9, 10]。在新型复合材料中,N 掺杂的 CQD 或氮/硫共掺杂的 CQD(N,S-CQD)表现出比原始复合材料高得多的荧光量子效率或光催化活性 [11, 12]。此外,N 掺杂 CQD 的性能增强与氮掺杂量呈正相关 [13, 14]。尽管这些研究令人信服地证明 N 掺杂显着影响 CQD 的性质,但是,关于 CQD 的有效异质掺杂方法的报道很少。由于掺杂剂无机前驱体的高溶解性,传统的水热碳化路线会导致反应溶液中残留大量掺杂剂,从而导致最终CQD中的N掺杂量很低。
在此,我们报道了仅在 50 秒内通过微波辅助方法合成富氮 N、S 共掺杂碳量子点(N、S-CQD)。选择柠檬酸 (CA) 作为碳源,硫脲不仅用作氮和硫源,而且用作弱碱。 N, S-CQDs 的氮和硫浓度分别达到 12.8 和 7.2 wt%,比 N-CQDs 和 N, S 共掺杂 CQDs 报道的高约 5 倍和 3 倍 [11, 14]。
方法
N, S-CQDs 通过以下方式获得:将 0.42 g (2 mmol) 柠檬酸一水合物和 0.46 g (6 mmol) 硫脲的混合物置于瓷坩埚中,并在微波反应器中加热 50 秒(445 瓦)。将得到的棕黄色产物加入30mL去离子水中形成黄色悬浮液,9000rpm离心20分钟。然后,用 0.22-μm 滤膜纯化上清液,并用去离子水通过透析膜(保留分子量,1000 Da)透析 24 小时。最后,透析液在真空下进一步冷冻干燥。以纯柠檬酸一水合物为原料合成原始CQDs,后续处理工艺与N,S CQDs相同。
在含有 1 μg mL −1 的 30 mL 柠檬酸-磷酸氢二钠缓冲溶液(pH ≈ 3.5,35°C)中测量 N, S-CQDs 对 H2O2 分解的酶模拟活性 N、S-CQDs 和 8 × 10 −4 四甲基联苯胺 (TMB) 底物的 M。在无色缓冲溶液中加入 160 μL H2O2 (30%) 溶液后,反应开始,然后每 2 分钟取出溶液在 652 nm 处测量 TMB 的蓝色氧化产物的吸光度。最后,计算氧化TMB的反应速率。 N, S-CQDs 的可重复使用性测试在含有 60 mL 柠檬酸-磷酸氢二钠缓冲溶液和 2 μg mL -1 的反应体系中进行 N、S-CQDs 以及 5 × 10 −3 M TMB 底物。在混合溶液中加入 H2O2 溶液(0.3%,320 μL)开始反应,1 小时后取少量溶液测量 652 nm 处的吸光度,完成第一个循环。然后,将 320 μL 新鲜的 H2O2 (0.3%) 溶液添加到反应系统中,用于下一个循环。在相同条件下重复其他三循环反应。减去最后的吸光度计算相应的吸光度。
透射电子显微镜 (TEM) 和高分辨率透射电子显微镜 (HRTEM) 图像是在 JEM-2100 电子显微镜上以高电压 (200 kV) 获得的。选区电子衍射 (SAED) 是通过 FEITF20(FEI 高分辨率场发射透射电子显微镜)在 200 kV 条件下测量的。紫外/可见吸收光谱用 UV-3600(岛津紫外-可见-近红外分光光度计)进行。荧光光谱在 F-7000(日立荧光光谱仪)上记录,电压为 700 V。荧光寿命和 FLQY 由 FM-4P-TCSPC(Horiba Jobin Yvon)测量。激发和发射波长分别为 358 和 436 nm。 X 射线粉末衍射仪 (XRD) 通过 D8 Advance(德国布鲁克 AXS Ltd.)使用 Cu Kα 进行表征 在 40 kV 和 40 mA 条件下。傅里叶变换红外 (FT-IR) 光谱使用 Nicolet iS10 (Thermo Fisher Infrared Spectrometer) 进行。 X 射线光电子能谱仪 (XPS) 在 PHI 5000 Versa (UIVAC-PHI) 上获得。 TG-MS(热重质谱)由 Netzsch STA 449C 测量,加热速率为 10 K min -1 在 N2 空气(10%,空气)流下,从 35°C 到 450°C 的最终温度。
结果和讨论
从 TEM 图像(图 1a)可以看出,所制备的 N、S-CQD 是均匀且分散良好的薄纳米片,平均直径为 2.0 nm。插入的 HRTEM 图像(图 1a)显示了非常清晰的 0.24 nm 晶格条纹间距,与石墨烯的(1120)面一致,表明 N、S-CQD 的晶核可能由石墨 sp 2 碳原子 [15, 16]。插入的 SAED 图像(图 1b)显示 N、S-CQD 是晶体,晶格边缘为 0.312 nm,对应于报告的图形 N-CQD [13]。这个d 值与所报道的具有石墨结构的 N、S 共掺杂 CQD 的 (002) 衍射面的晶面间距非常吻合 [11]。 N, S-CQDs 的 XRD 图显示了一个中心在 2θ 的宽峰 大约 25.5° 的值分配给石墨烯的衍射峰(图 1c),对应于 0.33 nm 的层间距 [17]。然而,已经报道了由尿素与柠檬酸钠或柠檬酸合成的 g-CNQDs 和 β-C3N4 [18, 19]。与我们的样品不同,g-CNQD 在 XRD 中在 27.4° 和 13.1° 处具有两个特征峰。 27.4° 处的强峰代表芳族系统的特征面间堆积,将石墨氮化碳标记为 (002) 峰,13.1° 处的弱衍射峰对应于标记为 (100) 峰的面间结构堆积基序。 g-CNQDs 是用高摩尔比 (6:1) 的 N/C 前体(尿素与柠檬酸钠)合成的 [18]。此外,更多的 N 注入核心以形成氮化碳点,热处理时间更长,达到 60 分钟。但我们的 N, S-CQD 样品的微波固相热解加热时间仅为 50 秒。与我们的样品不同的是,将尿素和柠檬酸溶液的混合物的体积煮沸至 100°C 并获得 β-氮化碳纳米晶。总之,我们推测 N/C 前驱体的低摩尔比、短反应时间和相对较高的温度可能导致石墨碳结构。与自由掺杂的 CQD [20] 相比,我们的 N、S-CQD 样品的 (002) 衍射峰从 23° 移至更高的角度 25.5°,这意味着层间距减小。 N、S-CQD 的类石墨烯层之间的强晶间电子堆叠相互作用。具有比碳原子更强的电负性,在共轭碳骨架中大量氮和硫原子的杂掺杂会导致整个共轭碳骨架的电子密度增加,从而缩短了面间距 [21, 22 ].
<图片>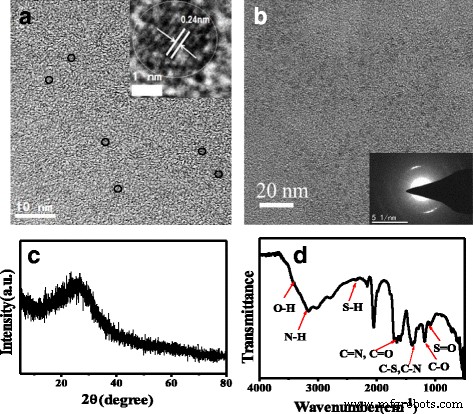
一 N, S-CQDs 的 TEM 图像(插入是 HRTEM 图像)。 b N、S-CQD 的 SAED 图像。 c XRD 和 d N, S-CQDs的FT-IR光谱
FT-IR 光谱(图 1d)证实了 N、S-CQD 的各种表面基团。 3163cm −1 处的波段 肩高 3416 cm −1 在 3000–3500 厘米 −1 范围分别代表 N-H 和 O-H 伸缩振动 [11]。这些相当大的氨基、羟基亲水基团可以使 N、S-CQD 具有优异的亲水性 [23]。出现在 1582、1656 和 1704 cm −1 附近的三重峰 可以分别分配给不同的特征振动键。 1704 cm −1 附近的峰值 是 C=O 羧基和 C=N 键的伸缩振动 [24],另外两个峰值在 1656 和 1582 cm -1 是酰胺基团伸缩 C=O(酰胺 I)的特征振动和 N-H 键(酰胺 II)的面内弯曲 [24, 25]。 1405 和 1345 cm −1 处的峰值 可以分别分配给 C-S 和 C-N 的振动 [17],而 1177 和 1084 cm -1 进一步证实 N、S-CQD 上存在 C-O 和 S=O 键 [17, 23]。 N, S-CQD 的 UV/vis 吸收光谱描绘了两个清晰的吸收带(图 2a)。 234 nm 处的强吸收带归因于芳族共轭体系 sp 的 π-π* 电子跃迁 2 域 [17],而 340 nm 处的弱吸收峰归因于 C=O 键的 n-π* 跃迁 [26]。注意到在 234 nm 处的吸收峰的相对强度比通过水热法合成的 N, S-CQD 样品强得多 [17, 26],表明形成了更多的芳香 sp 2 N 掺杂到共轭核心系统中作为吡啶 N 的域。此外,430 nm 附近的宽肩峰与 340 nm 处的峰重叠源于多种表面态跃迁 [26]。
<图片>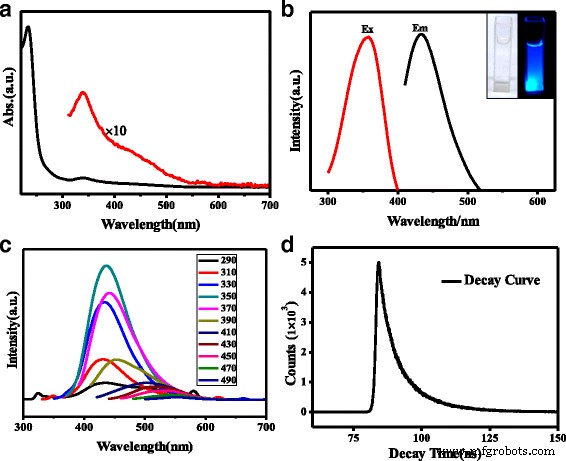
一 N, S-CQDs 的 UV/vis。 b N、S-CQD 和 插图 的 PL 是 N, S-CQDs 在环境光和 365 nm 照射下的图像。 c 具有不同激发波长的 N, S-CQDs 的 PL 光谱。 d 358 nm激发光下的光致发光强度衰减曲线
光致发光 (PL) 光谱(图 2b)说明 N、S-CQD 具有广泛的激发分布。由于 340 nm 的吸收峰,最大激发波长为 358(发射波长 436 nm)。从图 2b 的插图中可以看出,无色透明的 N, S-CQDs 水溶液在 365 nm 紫外线照射下变为亮蓝色。 N, S-CQDs 的溶液在 10 个月内保持清澈,没有沉淀; N, S-CQD 粒子的这种高稳定性归因于它们的尺寸小而均匀,以及表面的亲水基团。
N, S-CQDs 的光致发光量子产率 (PLQY) 在 358 nm 激发下计算为 23.6%,是 N 掺杂或 N, S-CQD 报道的结果的三倍 [20, 23]。相比之下,原始 CQD 的 PLQY 仅为 1.15%,远低于 N、S-CQD。据报道,CQDs 的 PLQY 与 CQDs 中的 N 掺杂量有关 [17, 27],因此,许多工作试图通过将反应时间延长至 19 来增加 CQDs 中的 N 掺杂量h 或将反应温度升高至 260 °C [11, 23]。然而,最终固体样品中的 N 掺杂量仍低于 6%。在我们的工作中,通过有效的固相微波辅助方式在 N, S-CQDs 上实现了 12.5% 的高 N 掺杂水平,其中柠檬酸和硫脲分子快速反应并避免升华。此外,硫脲作为弱碱可加快聚合速度。有趣的是,将硫脲和 CA 的比例从 3:1 改为 1:3 和 1:1,N、S-CQD 的 PLQY 分别略微降低到 7% 和 2.1%。此外,将反应时间增加至 2 分钟只会获得块状碳。为了进一步阐明反应机理,我们获得了纯 CA 和 CA 与硫脲的反应物混合物的热重-差热分析仪 (TG-DTA) 和 TG-质量曲线。如图 3a 所示,在纯 CA 的 TG-DTA 曲线中可以观察到三个放热峰;第一个峰对应于吸收水,包括结晶水。 154°C 处的第二个尖峰与 CA 晶体的熔化放热有关,而以 214°C 为中心的宽峰与分子间脱水和碳化有关。而对于 CA 和硫脲的混合物,在 TG-DTA 光谱上可以观察到后两个放热峰的很大变化(图 3b),第二个放热峰出现在 118°C 的低温下,表明酸-CA 和硫脲之间的碱相互作用导致熔化放热步骤中 36°C 的急剧下降。此外,除了对应于脱水和碳化的第三个放热峰外,在 214 和 236°C 处有两个峰,在 170°C 处可以观察到一个弱峰,这意味着硫脲的加入可以促进脱水和碳化过程.比较纯 CA 和 CA 和硫脲混合物的 TG 质谱(图 3c),可以发现 H2O 释放峰的最高温度从纯 CA 的 215°C 降低到 180°C CA和硫脲的混合物。类似地,CA 的 CO2 释放峰值的最大值在 227°C,但它转移到低温并分别在 179 和 198°C 分两步释放 CA 和硫脲的反应混合物。脱水和碳化过程中的这种温度降低与 TG-DTA 结果非常吻合,这意味着这两个系统的反应方法不同。对于纯 CA,分子间脱水和碳化在高温下同时发生。而对于CA和硫脲的混合物,CA的羧基和硫脲的氨基之间首先发生分子间脱水反应,然后逐步碳化形成N,S-CQDs的碳核。与CA分子之间的弱氢键相互作用相比,羧基和氨基之间的强酸碱相互作用导致脱水温度显着降低。有趣的是,如图 3c 所示,纯 CA 和 CA 与硫脲的混合反应物的残余质量分别为 1 和 21 wt%,这表明添加的硫脲可以起到弱碱的作用,降低避免升华,从而提高N, S-CQDs中N-和S-掺杂的含量,并获得高产率。
<图片>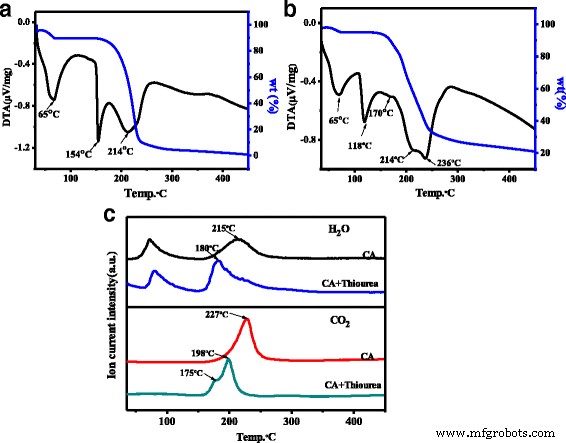
a的TG和DTA曲线 纯柠檬酸,b 柠檬酸和硫脲的反应混合物。 c 纯CA与柠檬酸和硫脲混合反应物的TG-Mass曲线
图 2c 说明了具有不同激发波长的 N、S-CQD 的发射光谱。当激发波长从 290 纳米变为 370 纳米时,440 纳米处的发射峰几乎没有变化。发射分量的能量相当恒定,很可能源自 340 nm 处 n-π* 跃迁的吸收。已经通过将复杂的发射峰拟合到多个高斯函数来研究 CQD 的与激发无关的发射特性,并推导出类似的结论 [26]。而当激发波长从 390 到 490 nm 变化时,PL 发射光谱随着激发波长的增加表现出红移,表征激发波长相关的特性。这可以归因于 C=O 的各种表面状态或酰胺基团作为离散激子俘获中心的作用,以影响 PL 过程中的发射能量 [11, 19, 28]。多分散性和表面异质性是激发波长相关 PL 行为的起源 [28, 29]。 430 nm 附近的宽吸收峰是各种表面状态的集合,包括羧基和酰胺,这使 N、S-CQD 的激发波长依赖于 PL 行为成为可能。确定 N, S-CQD 的荧光寿命以评估其光学性质(图 2d)。 N, S-CQD 样品的 PL 衰减曲线可以用双指数公式拟合,其中 τ 1 是 3.48 ns,τ 2 为 11.05 ns,平均寿命为 6.72 ns。与原始 CQD [30] 的平均寿命 2.42 ns 相比,τ 的荧光寿命显着更长 1 和 τ 2 是在我们的样本上获得的。据报道,τ 2 比例和平均寿命随着 N 掺杂量的增加而变长,得出的结论是 τ 2源于表面态[11, 31]。
XPS 证实了 N, S-CQDs 的形成。如图 4a 所示,O 1s 在 530、399、284、222 和 164 eV 处有五个不同的峰 , N 1s , C 1s, S 2s 和 S 2p 信号分别表明 N 和 S 确实被注入到 CQD 的框架中 [17]。高分辨率 C 1s XPS 光谱(图 4b)显示了 C 结构的三个特征,包括芳香共轭 sp 2 C (C=C) 在 284.4 eV,sp 3 C(C–N、C–O、C–S)在 285.6 eV,C=O/C=N 在 288.1 eV [11]。 N 1s N、S-CQD 的 XPS 光谱(图 4c)在 399.5、400.3 和 401.0 eV 处显示三个峰,分别代表吡啶 N、吡咯 N 和酰胺 N [17, 24]。在 g-CNQD 中,从实验 XPS 强度比得出的原子比 Ncore/Ccore 等于 1.40,接近 C3N4 [19] 的预期值 1.33。我们对我们的样本进行了类似的数据分析,取了 285.6 eV 的 C 1s 峰值为“Ccore”,而 N 1s 的峰值为 400.3 eV(吡咯)和 399.6 eV(吡啶) 作为“Ncore”(因为结合能值与 [19] 中 NCore 的 399.9 eV 相似),计算出的 Ncore/Ccore 为 0.43,远小于 C3N4 的 1.33。此外,发现我们的 N, S-CQD 中吡啶 N 与吡咯 N 的相对比例与通过水热法合成的 N 或 N, S 共掺杂 CQD 的比例有很大不同 [17, 21]。吡啶 N 是我们的 N, S-CQD 样品中的主要掺杂剂,是吡咯 N 的 1.5 倍,但对于许多热合成样品,它通常小于 1.0。如此高的吡啶氮可能赋予 N, S-CQDs 优越的性能,用于进一步的催化应用,因为它们可以作为催化活性位点 [32]。此外,边缘的吡咯氮是表面缺陷的重要组成部分,可以作为光致发光中心 [17, 27]。 S 2p XPS 光谱(图 4d)显示了 163.3 和 164.4 eV 处的两个典型信号,它们对应于 S 2p 3/2 和 S 2p 分别为噻吩 S 的 1/2 [16]。结合FT-IR光谱,我们推测硫原子以噻吩S的形式成功掺杂到N, S-CQDs的骨架中,并存在于N, S-CQDs的边缘以提高其PLQY。
<图片>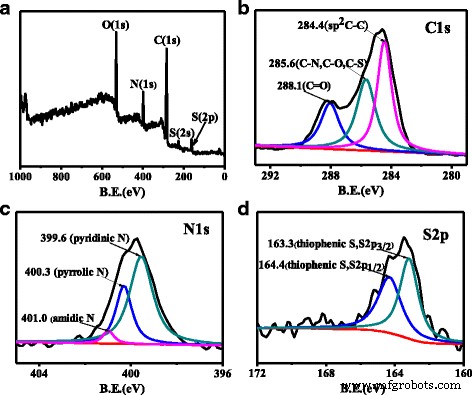
一 N、S-CQD 的全扫描 XPS。 C 1s的高分辨率XPS b N 1s c 和 d S 2p N, S-CQDs的光谱
酶催化因其高特异性和活性而备受期待。辣根过氧化物酶 (HRP) 是研究最多的植物酶,它含有血红素组中卟啉循环的活性中心,可有效催化单电子氧化,过氧化氢对多种有机和无机底物进行氧化 [33, 34]。通过测量蓝色氧化产物的吸收, 测试了富含吡啶的 N, S-CQDs 在 H2O2 存在下氧化 3, 3', 5', 5'-四甲基联苯胺 (TMB) 过氧化物酶底物的模拟特性652 nm 处的 TMB。 N, S-CQDs 的 UV/vis 吸收在 234 和 340 nm 处达到峰值。图 5a 说明了在 N、S-CQD 和原始 CQD 存在下,TMB 衍生的氧化产物浓度 (μmol/L) 随时间变化的拟合线。反应速率 (r ) 对于 H2O2 在 N 上的分解,作为酶模拟物的 S-CQDs 为 2.16 × 10 −3 μmol −1 L −1 S −1 ,这是相同条件下原始 CQD 和先前报道的无掺杂 CQD 的两倍 [35, 36]。富含吡啶的 N, S-CQDs 的优异活性可归因于 N 的高掺杂含量,该 N 具有比碳原子大的电负性,以增加 N, S-CQDs 的电子密度,尤其是占主导地位的吡啶 N 拥有一个孤对电子导致 N、S-CQD 的 π 共轭框架中的电子密度和迁移率增强,从而加速反应。这是首次报道CQDs的过氧化氢酶模拟性能依赖于吡啶N在碳骨架中的主要掺杂。
<图片>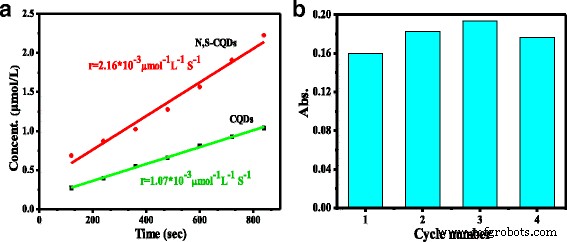
一 N、S-CQDs 和自由掺杂的 CQDs 的初始反应速率。 b N,S-CQDs的复用性测试
通过连续四次使用过氧化氢酶模拟反应来研究 N, S-CQD 的可重用性(图 5b)。在四次循环使用中,未观察到 N, S-CQDs 的活性明显下降。 N, S-CQDs 固有催化活性的高稳定性归因于 C=C 骨架中主要的吡啶 N 掺杂,因为吡啶 N 可以作为 H2O2 分解的有效酶模拟催化位点。
结论
总之,我们通过微波固体聚合法在短短 50 秒内合成了具有高 N 和 S 掺杂水平的富含吡啶的 N、S-CQD。硫脲不仅作为 S 源,而且作为弱碱加速低温下的分子间脱水和多步碳化,这使得 N、S-CQD 中的高 N-和 S-掺杂水平和主要的吡啶 N 注入共轭框架作为酶模拟活性位点。该工作为合成具有高PLQY和酶模拟活性的富含吡啶的N,S-CQDs提供了一种有效的方法。
纳米材料
- DNA 合成
- S、N 共掺杂石墨烯量子点/TiO2 复合材料用于高效光催化制氢
- 高效独立激发蓝色发光碳点
- 重吸收抑制的 II 型/I 型 ZnSe/CdS/ZnS 核/壳量子点的合成及其在免疫吸附测定中的应用
- 退火 GaAsBi/AlAs 量子阱中的铋量子点
- InP/ZnS 核/壳量子点的绿色合成在无重金属发光二极管中的应用
- 磁性碳微球作为可重复使用的吸附剂从水中去除磺胺
- 从豆腐废水中合成荧光碳量子点的简单方法
- 水溶性硫化锑量子点的合成及其光电特性
- 石墨烯/Ag3PO4 量子点复合材料的简便一步声化学合成和光催化性能
- 掺杂铕的核-壳 ZnSe/ZnSe 量子点中粒子间能量转移的壳厚度依赖性
- 1.3–1.55-μm 窗口中变质 InAs/InGaAs 量子点的带间光电导率