

限制对 PMMA 矩阵中 P3HT 链的光物理特性的影响
摘要
嵌入聚(甲基丙烯酸甲酯)(PMMA)基质中的聚(3-己基噻吩)(P3HT)链的排列对形成的 P3HT 聚集体的光物理性质(如电子吸收光谱、带隙和光致发光量子产率)的影响具有被研究过。已经发现 PMMA 基质中 P3HT 分数从 25% 到 2% 的变化伴随着光致发光量子产率的增加、带隙的红移和 P3HT 微晶的结构变化。上述变化伴随着 P3HT 部分的连续网络被破坏成更小的 P3HT 颗粒,尺寸范围从几微米到几十纳米。结果解释为分子间堆积的变化和分子内扭转紊乱的减少。讨论了对上述变化的最大贡献来自P3HT簇和PMMA环境界面处的P3HT分子。
背景
在过去十年中,塌陷线圈和共轭聚合物纳米级受限系统的光物理学引起了相当大的兴趣 [1,2,3,4]。特别是,聚(3-己基噻吩)(P3HT)纳米级聚集体和微晶中的激子产生、辐射复合和光生电荷转移过程对有机太阳能电池的性能有直接影响,其中该聚合物用作活性成分。结果表明,孤立的 P3HT 分子和 P3HT 聚集体的发射性质是不同的。分子发射通常源自常见的链内激子状态,对应于具有减少的扭转紊乱的松弛链 [5]。 P3HT 聚集体的发射光谱也源于一个共同的发射状态,但对应于通过单个或多个能量转移步骤下降到最低能量域的链间单线态激子 [6]。由于链间离域和激子在凝聚材料中的耗散,与溶液中的自由分子相比,P3HT 薄膜中有序层状结构的光致发光 (PL) 的量子产率 (QY) 受到强烈抑制 [7]。另一方面,QY 可以通过控制温度 [8] 或 P3HT 链的区域规整性 [9] 来增强。例如,结果表明,与区域随机 P3HT 薄膜相比,区域规则 P3HT 薄膜具有更弱的光学跃迁,因为与区域随机 P3HT 中激子的链内特征相比,薄片中最低激子的链间贡献更大 [9]。因此,开发简单有效的策略来通过改变共轭大分子的分子内设计和分子间组装和排序来操纵其光学性质,对于进一步了解这类有趣的材料以及它们在有机电子学中的广泛应用具有巨大的潜力。
这项工作的目标是展示 P3HT 链的排列变化如何影响物理特性,例如 P3HT 纳米级粒子的电子吸收光谱、带隙和发射 QY。使人们能够调整共轭聚合物薄膜的光物理特性的一种有前途的策略是与另一种惰性聚合物混合。众所周知,在 P3HT 的情况下,它的光学特性很容易受到合适的宿主介质的影响。例如,李等人。表明 P3HT 纳米粒子的吸收和发射实验中的光学跃迁能受到去离子水在高达 150 °C 的温度下在高压釜中进行水热(极性)处理的影响 [10]。赫尔曼等人。表明 P3HT 与极性聚(环氧乙烷)(PEO)的混合导致 0-0 振荡器强度增加以及光学吸收光谱的显着偏移 0.1 eV [11]。此外,Kim 等人。在将 P3HT 和 PEO 混合并从极性溶剂混合物中纺丝后,电纺 P3HT 纳米纤维的光学特性发生了类似的变化 [12]。其他研究表明,通过与聚(乙二醇)混合而无需额外的极性溶剂添加剂 [13],P3HT 薄膜的光吸收光谱会发生轻微的红移。因此,上述实验表明 P3HT 的光物理特性可以很容易地通过处理手段进行操纵。尽管上述研究表明宿主环境对 P3HT 聚集体的带隙有显着影响,但发射 QY 的变化很少受到关注。例如,Kanemoto 等人。已经表明,通过使用中等惰性聚合物(如聚丙烯)进行稀释,可以提高固态共轭聚合物的 PL [14]。然而,这种效果是通过将聚集体转化为共轭聚合物的分子形式来实现的。
在这里,我们证明了共轭聚合物 P3HT 与极性聚甲基丙烯酸甲酯 (PMMA) 的混合,其中形成了微米级和纳米级的 P3HT 颗粒,诱导了 P3HT 聚集体物理特性的系统变化。我们表明,随着 P3HT 与 PMMA 的重量比降低,P3HT 分数表现出带隙红移、排序的改善和发射 QY 的增强。我们表明,这些变化很可能是由于在主体材料疏水力的作用下,共轭聚合物骨架在 PMMA 存在下的平面化。
方法
样品准备
区域规则 P3HT(~ 93% RR,99,995% 痕量金属基础,数均分子量 (Mn) 在 15-45 kDa 范围内,Sigma-Aldrich)的初始储备溶液在氯苯中制备,浓度为 1.0 wt%( CB)。通过将必要量的聚(甲基丙烯酸甲酯)(PMMA,平均分子量(Mw)为 120 kDa,Sigma-Aldrich)添加到 CB 中的 P3HT 溶液中,然后在超声浴中处理,制备 P3HT 和 PMMA 的二元混合物30 分钟。通过在玻璃基板上以1500 rpm、30 s的速度旋涂制备薄膜。
对于透射电子显微镜 (TEM) 研究,用丙酮将薄膜刮到容器中,然后停留几个小时以确保所有 PMMA 完全溶解,释放出几乎不溶于丙酮的 P3HT 聚集体(P3HT 在丙酮中的溶解度较低)低于 0.1 毫克/毫升 [15])。将少量溶液滴铸到 TEM 碳网格上,然后蒸发丙酮。丙酮中的 PMMA 溶液滴铸在单独的网格上,以获得整洁的 PMMA 样品的图像。
光谱测量
使用 SPECORD M40 和 OLIS Cary 14 双光束分光光度计测量吸收光谱。裸玻璃板作为参考。使用 SPEX Fluorolog 1680 双光谱仪收集荧光光谱,氙灯作为光源。激发波长选择在 468 nm。为了比较它们的光谱特征,吸收光谱在下面给出,归一化到它们的最大值,并且 PL 光谱针对配准系统的灵敏度进行了校正,并归一化为激发波长下的样品吸收,即,显示了 PL 光谱样品发射的相对QY。
瞬态吸收 (TA) 泵浦探针测量是使用钛蓝宝石激光系统进行的。激发波长设置为 410 nm。 TA 测量是使用泵(重复率为 1 kHz,脉冲持续时间为 ~ 100 fs)和由蓝宝石晶体产生的白光连续谱作为探针进行的。泵浦光束以 CPA 系统重复率的一半 (500 Hz) 进行机械调制,并且 ΔT /T 或 ΔOD 是通过使用锁定放大器的相位敏感技术检测到的。泵浦光束的偏振相对于探测光束的偏振角为魔角 (54.7°)。测量的分数传输信号,即 TA,由 TA =-ΔT 给出 /T =(T on-T 关闭)/T 关闭,其中 T on 表示带泵的探头传输,T 在泵关闭的情况下关闭探头传输。所得光谱经波长校准程序校正。
显微镜测量
通过光学显微镜和 TEM 研究样品的形态。使用配备有照相机和计算机的光学显微镜 ULAB XY-B2 拍摄样品的光学显微照片。 TEM 研究使用 JEOL JEM-1400 仪器在 80 kV 下运行。
结果
光物理研究
P3HT 复合薄膜的电子吸收光谱(图 1a)表明吸收从大约 ~ 650 nm (1.9 eV) 开始,对应于 P3HT 晶体的带隙,随后是 ~ 605、560 和 525 nm 的振动复制它们分别与基本 (0-0) 转换、(0-1) 和 (0-2) 边带相关。随着 P3HT 与 PMMA 的重量比降低,光谱逐渐演变。首先,(0-0)与(0-1)吸收的幅度比增加。其次,吸收光谱从光谱的短波长侧开始变窄;该区域通常归因于无定形状态下无序分子的吸收,因为在约 460 nm 附近观察到 P3HT 在稀释溶液中的分子吸收;因此,上述变化表明样品中无序无定形部分的减少 [6, 8]。第三,与(0-0)跃迁相关的最大吸收从602nm逐渐移动到608nm;从吸收边切线与横坐标轴的交点计算的带隙也随着复合薄膜中 P3HT 与 PMMA 的比率降低而从 1.92 eV 红移至 1.89 eV。
<图片>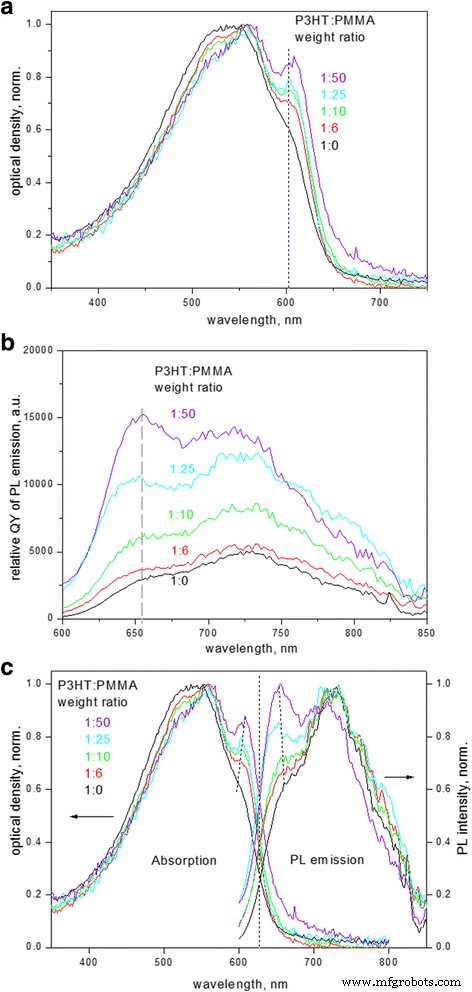
一 归一化电子吸收光谱,b PL 光谱(相对 QY)和 c 不同P3HT:PMMA重量比的P3HT-PMMA复合薄膜归一化吸收光谱和PL光谱对比
PL 发射光谱(图 1b)证明斯托克斯位移约0.15 eV,并且光谱具有与吸收光谱相似的行为,具有边带的镜像序列。 PL 光谱的形状,特别是 (0-0) 与 (0-1) 带强度的比率也取决于 P3HT 在 PMMA 基质中的分数。上述变化与电子吸收光谱和 PL 光谱密切相关,并表明 P3HT 薄膜的有序程度 [16, 17]。 (0-0) 到 (0-1) 的幅度比小于 1,是 H-聚集体与非聚集链序列共存的特征 [18, 19]。此外,随着 P3HT 与 PMMA 的比率降低,薄膜的吸收光谱和 PL 光谱都显示出相对于边带的第一个最大值的强度增加(图 1a,b)。光谱中 (0-0) 跃迁强度的相对增加证明有利于薄膜中 P3HT 链的重排。 (0-0)与(0-1)波段的强度比与自由激子带宽W有关 同样,其非零量级反映了聚合物链中的无序程度 [16] 并且可以通过使用方程计算。 (1) 下面假设一个 Huang-Rhys 因子等于 1 [20, 6]:
$$ \frac{A_{0-0}}{A_{0-1}}\approx \frac{n_{0-1}}{n_{0-0}}{\left(\frac{1-\压裂{0.24W}{E_p}}{1+\frac{0.073W}{E_p}}\right)}^2 $$ (1)其中 n 0−i 是 0–i 峰值和 Ep 处的折射率的实部 是耦合到电子跃迁的主振荡器的声子能量。在方程式中。 (1), 折射率比为∼ 0.97 [6], 主要分子内振动E p 以 0.18 eV 的 C=C 对称拉伸为主 [21]。在更有序的聚合物链中,库仑链间耦合较弱,这导致激子带宽变窄。非零激子带宽也影响 P3HT 中第一个吸收最大值的能量位置,因为激发发生在激子带的上能级,而发射分别发生在能带的低能级。影响如下:激子带宽越宽,第一个 PL 发射最大值(指定为 P3HT 聚集体中的 0-0 跃迁 [20])和第一个吸收最大值的间隔越大;这种趋势由图 1c 中的虚线表示。
随着 P3HT 与 PMMA 的比率降低,激子带宽表现出变窄(图 2a),伴随着 P3HT 发射的 QY 增加四倍(图 2b)。一方面,这种行为应该对应于 P3HT 链排序。另一方面,激子带宽的降低与链内相关性的增加和链间相关性的降低有关 [21],这意味着聚合物主链的链内顺序和共轭长度同时增加,参与 π 的链数量减少- 激子离域的π相互作用,理论上接近理想有序长链的零激子带宽[22]。然而,图 2a 中激子带宽对 PMMA:P3HT 比的依赖性可以通过具有偏移的指数拟合。偏移量表示带宽没有接近于零;相反,它达到了 45 ± 5 meV 的饱和水平(图 2a)。这意味着 P3HT 矩阵中有序 P3HT 聚集体的非零大小有限制,这会产生链间激子,而与 PMMA 矩阵中 P3HT 的分数无关。这一发现与区域规整聚噻吩的自组装 [23] 和形成高度有序的结晶域的强大特性有关,其特征尺寸可以小至~ 10 nm [24, 8]。然而,我们计算出的激子带宽极限值比从其他不良溶剂(如均三甲苯或异丁二烯(W ~ 30 meV [21]);在我们的案例中,这种差异可以通过在三元 P3HT-PMMA-CB 体系 [25] 的成膜过程中存在良好的溶剂,即 CB 来解释。
<图片>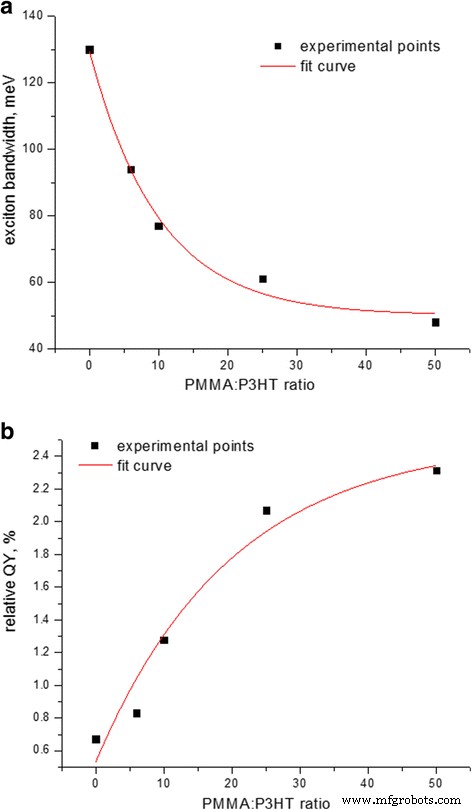
一 激子带宽和b P3HT-PMMA 复合薄膜发射的相对 QY 与 PMMA:P3HT 重量比的函数关系,假设纯 P3HT 薄膜的 QY 约为 0.5% [25]
TA 光谱为 PMMA 基质中 P3HT 聚集体的排序提供了额外的证据。纯 P3HT 和 P3HT:PMMA 复合薄膜的 TA 光谱比较如图 3 所示。典型的光谱由两个负带组成,其中一个是 530-630 nm 区域的基态漂白 (GSB),指示 P3HT 的 0-1 和 0-0 吸收的漂白,而在~ 700 nm 处的另一个带指示受激发射 (SE)。光谱中~ 660 和~ 950 nm 处的正带是极化子吸收的特征,分别在有序晶体域中离域和在无序非晶域中局部化 [25,26,27]。 ~ 1200 nm 处的带通常分配给 P3HT 中的单线态激子 TA [7, 28,29,30]。上述光谱(图 3)中的特征差异在于 PMMA 基质中的 P3HT 链在~ 660 nm 处表现出明显的离域极化子吸收,表明存在大量的 P3HT 结晶有序区域,而在~ 700 nm 处显着的 SE整洁的P3HT薄膜具有无序P3HT链中链内激子的特征[8]。
<图片>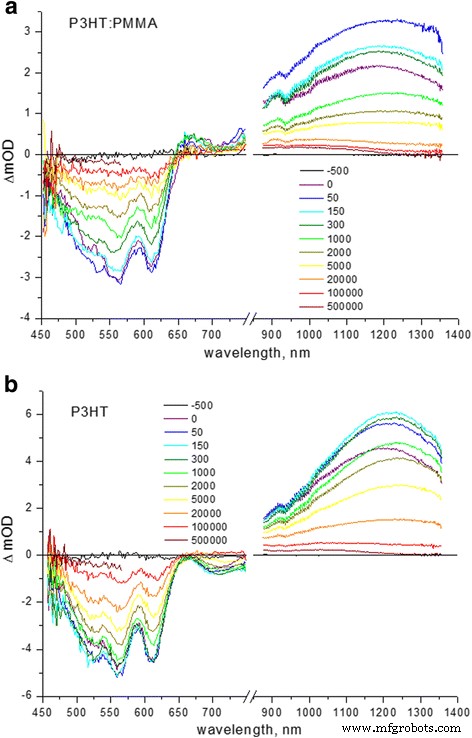
a的TA谱 P3HT:PMMA(1:50 重量比)和 b 整洁的 P3HT 旋涂薄膜。时间延迟在竖条中以飞秒表示
TA 光谱中 (0-0) 跃迁和 (0-1) 振动边带的分析揭示了纯 P3HT 和复合 P3HT-PMMA 薄膜中不同的相对弛豫动力学(表 1)。在纯 P3HT 薄膜中,与 (0-0) 跃迁的弛豫相比,(0-1) 振动边带衰减更慢,表明 (0-1) 的主要寿命(贡献 ~ 60%)为 7.0 ps转换与 (0-0) 转换分别为 5.3 ps。在 P3HT-PMMA 复合材料中,(0-1) 和 (0-0) 跃迁的主要寿命 (~ 73%) 较短且相似,为~ 1.8 ps,而次要组分的寿命 (~ 27%) (0-0) 跃迁较慢(分别约为 300 与 200 ps),从而提供了(0-1)振动边带整体的更快弛豫(图 3)。皮秒级弛豫的快分量是扭转弛豫的特征,导致光激发时 P3HT 链平面化 [31, 32],而慢分量是 TA 测量探测到的非荧光激子寿命的特征 [7] .纯 P3HT 和 P3HT-PMMA 复合薄膜在 GSB 区域的不同弛豫行为表明,光激发后复合样品中 P3HT 链的平面化速度更快;这意味着链在基态下已经部分平面化,即它们在复合膜中的扭转无序比在纯 P3HT 膜中少。另一方面,激子 TA 在 P3HT-PMMA 复合材料中也表现出更快的衰减,典型时间为 0.6 ps (68%) 和 19 ps (32%),而 2 ps (51%) 和 40 ps (49%) ) 分别为纯 P3HT 薄膜(表 1)。上述衰变的超快成分可归为链内激子能量从高能位点向低能位点的转移 [33] 以及发生快速激子迁移后等能能量转移的较慢成分 [34]。似乎合理地表明链内激子迁移在更有序的链中进行得更快,没有扭转无序,再次证实 P3HT 链在复合样品中具有更好的有序性。
形态学研究
随着 P3HT:PMMA 比率的变化,显微镜研究使我们能够观察 PMMA 基质中 P3HT 部分的尺寸分布。首先,复合 P3HT:PMMA 薄膜显示出高度结构化的形态,指出 P3HT 和 PMMA 发生相分离,与形态相对光滑的纯 P3HT 薄膜形成对比(图 4)。然而,在相对高浓度的 P3HT(~ 10 wt% 或更高)下,P3HT 部分是连续的,并在 PMMA 基质中形成渗透网络。在低浓度的 P3HT 下,聚合物部分转变为分离微米和亚微米尺寸的 P3HT 颗粒。 TEM 图像(图 5a)显示颗粒可以小到 ~ 30 nm,并且实际上具有理想的球形。颗粒的球形使我们能够提出 P3HT 和 PMMA 之间的排斥力,其中 P3HT 的无定形(“液体状”)相倾向于从 PMMA 基质分离成具有最小表面的致密颗粒。因此,在 P3HT 颗粒中应建议有足够比例的非晶相。选区电子衍射 (SAED) 图案(图 5a,右侧)显示粒子的无定形和结晶特征的叠加。在样品中也发现了单独的高度结晶的非球形区域(图 5b)。因此,建议球形 P3HT 颗粒由被 P3HT 非晶相包围的结晶核组成。拉希米等人。发现即使是高度有序的 P3HT 单晶也被一小部分约 12% 采用溶液状构象的分子包围,并且非晶层的特征厚度约为 10 nm [35]。假设在结晶核周围形成了类似厚度的非晶层,很容易理解,由于这种非晶壳,具有~ 30 nm大小的颗粒可以很容易地采用球形。
<图片>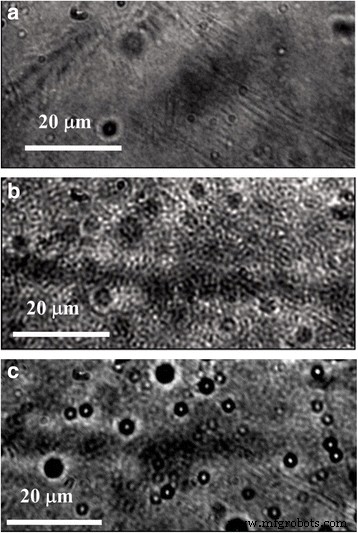
a 的光学显微照片 整洁的 P3HT 和 b , c P3HT:含 b 的 PMMA 薄膜 10 wt% 和 c PMMA中2wt%的P3HT
<图片>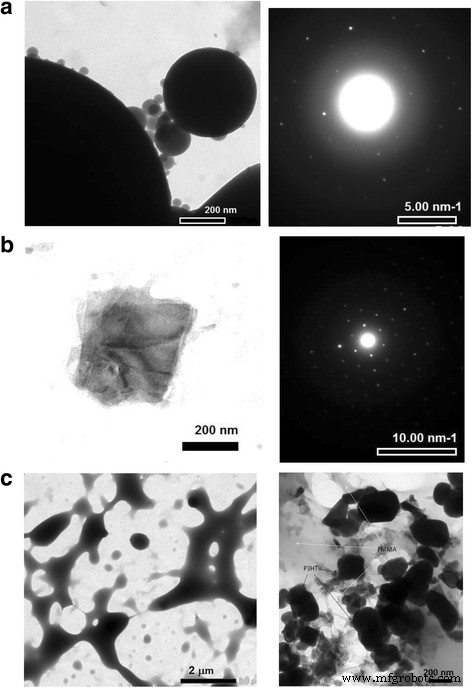
a 的 TEM 图像 P3HT的球形颗粒,b P3HT 的结晶域,以及图像右侧相应的 SAED 模式; c 整洁的 PMMA(右图 ) 和 P3HT-PMMA 混合(左图 ) 进行比较
晶体结构的赋值
一般来说,热力学有利的纹理被假定为由 P3HT 的边缘取向链形成 [36, 37]。这种结构是在接近平衡的条件下获得的,这些缓慢的成膜方法如滴铸 [38, 39]、浸涂和从高沸点溶剂中旋涂 [40]。在 TEM 网格上制备的样品中,由于疏水网格表面(碳)更喜欢与 P3HT 分子的烃取代基相互作用,因此优选边缘取向。这种取向提供了 SAED 图案中 P3HT 片晶主要 (010) 和 (001) 面的“可视化”(图 6)。
<图片>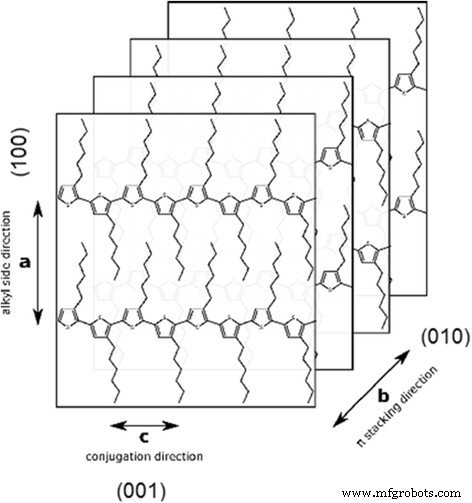
P3HT的晶体结构示意图
连续聚噻吩骨架沿 b 的以下堆积周期 获得的轴:球状结构为 0.45±0.1 nm(图 5a,图 7),分离薄片为 0.48±0.1 nm(图 5b)。考虑到 a 和 b 轴通常接近 90° [41]。此外,一些 SAED 图案使我们能够找到 (h00) 衍射环(图 8),从那里可以确定堆叠间距离(由 n 层分隔的 P3HT 链之间的距离) -己基侧链,即沿着 a 单斜晶胞的轴,图 6),为 1.23 nm。获得的距离是晶型 II 的特征。因此,如果我们试图将晶体归因于形式 II,我们应该考虑到形式 II 晶体在 b 之间具有倾斜角 γ =68° 轴和噻吩平面 [42],从中可以计算出短晶面间距分别为 0.417 和 0.445 nm。后一个值与晶型 II (0.44 nm [43]) 中的短晶面间距非常吻合,而前者更好地对应于中间晶型 I',晶面间距为 0.41–0.42 nm [44]。
<图片>
一 TEM 图像和 b P3HT-PMMA(1:50重量比)共混样品中球状P3HT域的SAED
<图片>
P3HT-PMMA(1:50重量比)共混样品中P3HT结构域的SAED
讨论
这项工作的主要发现是,P3HT 凝聚相的发射 QY 可以增强,这不是由于紧密堆积的聚集体解缠结,相邻分子将大量激子淬灭成 P3HT 的分子形式,而是通过简单地减小 P3HT 凝聚相的尺寸相到微米和纳米粒子。可以考虑导致上述现象的两个主要原因:首先,P3HT / PMMA 的界面面积增加,其中界面分子由于表面积与体积比的增加而增加了它们对发射特性的贡献。 P3HT颗粒;其次,由于PMMA的排斥力,P3HT链在结晶域中的排列发生了变化,随着P3HT与PMMA的比例降低,P3HT分子会受到影响。
第一个可能的原因暗示了 P3HT 环境介电常数的变化。事实上,胡等人。据报道,用具有低介电常数(ε <3)的非极性溶剂替代具有高介电常数(ε> 3)的相对极性溶剂导致 P3HT 聚集体的荧光 QY 增强几乎一个数量级 [45]。应该注意的是,PMMA 具有 ε> 2.8 [46],原则上可以认为它会影响 PL 发射的 QY。为了验证这个因素的贡献,我们检查了 P3HT 的 PL 发射,因为溶剂环境逐渐被 PMMA 分子取代(图 9)。在第一个实验中,将相同数量的 P3HT 储备溶液分别添加到含有 CB 的比色皿和 PMMA 在 CB 中的浓缩溶液中,其中溶液体积相同(图 9a)。在第二个实验中,为了消除供应 P3HT 溶液的注射器体积可能出现的偶然误差,将 PMMA 粉末添加到 P3HT 溶液中,并在 PMMA 溶解过程中连续测量光谱(图 9b)。两个实验都显示在 PMMA 存在下 P3HT 溶液的相对发射 QY 有小幅但明显的增加。因此,CB (ε ~ 5.6) 的介电常数到 CB-PMMA 混合物的介电常数的变化,然后是薄膜中纯 PMMA 环境的变化应该有助于增强荧光 QY。然而,溶液中的这种影响被评估为很小,导致 PL 的 QY 仅增加 ~ 14%。另一方面,在薄膜中,发现 PL 的 QY 增加高达 ~ 400%(图 2b)。因此,介电常数的相对变化对复合薄膜中PL的QY的提高只有微弱的影响。
<图片>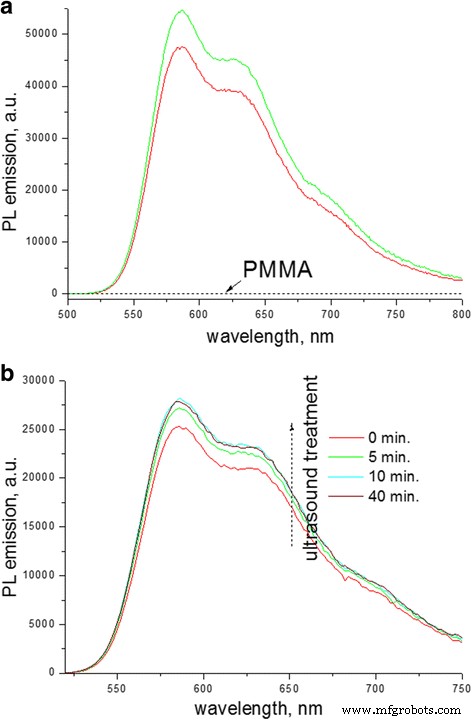
P3HT 溶液 (0.01 wt%) 在 CB 中的 PL 发射光谱 (λexc =468 nm):a 之前(红色 ) 和之后 (绿色 ) 与 PMMA (5.4 wt%) 溶液混合,也给出了 PMMA 的 PL 光谱; b 将 PMMA 粉末添加到纯 P3HT 溶液中后(红色 ) 然后在超声波浴中连续处理比色皿并逐渐溶解 PMMA 至 ~ 3.25 wt%
从 P3HT-PMMA 薄膜的光谱变化中可以特别推断出的另一个重要因素是 PMMA 基质中 P3HT 域中聚合物链的相互排列的变化。应该注意的是,P3HT 晶体可以采用不同的形式,即退火后在薄膜中最常见的形式 I [47],或代表能量更稳定情况的形式 II [42]。例如,可以通过亲水性聚合物基质和不良溶剂(如水)在成膜过程中在 P3HT 链上的协同作用获得形式 II [11],并且它在吸收光谱中显示出显着的红移 [35];在我们的结果中观察到类似的趋势,显示带隙从 1.92 到 1.89 eV 的红移(图 1a)。有趣的是,与 I 型的堆叠距离在 0.340 和 0.414 nm 之间 [48,49 ,50]。同时,形式 II 中存在更紧密的烷基侧链交叉,链间距离(在烷基指向的方向上)为 1.20 至 1.31 nm [42],而链间距离约为 1.55 至 1.73 nm晶型 I [48, 50] 中的纳米;更紧密的交叉似乎更好地稳定了II型晶体的链内排序。
上述关于 P3HT 不同晶型的讨论对于理解在不同 P3HT 与 PMMA 重量比下在 PMMA 基质中形成的 P3HT 晶体的结构转变很重要。首先,已经发现在旋涂 P3HT-PMMA 薄膜中与(0-0)带相关的最大位置在小的 P3HT 与 PMMA 比率下经历了轻微的红移,即从 602 到 608 nm(图 1a) .其次,显微镜研究使我们能够区分混合样品中的两种类型的晶体,它们在堆叠方向(沿着 b P3HT 晶体的轴)分别为 0.417(球状结构的特征,见图 5a 和图 7)和 0.445 nm(片状结构的特征,如图 5b 所示)。后一个值与上面讨论的晶型 II 非常吻合,而前者更好地对应于 Roehling 等人报道的中间体 I’。 [44], which possesses the interplanar distance of 0.41–0.42 nm. They also showed that the form I’ demonstrates an increase in the coherent domain size in the π − π stacking direction by a factor of ~ 2 (from 6.3 to 12.4 nm), as compared to form I in samples prepared from p-xylene, which can be responsible for the enhancement of the (0-0) band relatively to the (0-1) band in P3HT samples [50].
Based on the above discussion, we can conclude that the composite P3HT-PMMA samples contain crystals of both forms of P3HT (I’ and II) because the interchain stacking distance varies from 0.42 to 0.44 nm for crystals of the different morphology. Thus, it can be suggested that the changing (0-0) to (0-1) ratio is related to the changing weight ratio of the different crystalline forms of P3HT, respectively, and the increasing (0-0) to (0-1) ratio most probably is due to increasing fraction of the form I’ in the blend, which promotes the increasing coherent domain size in the π − π stacking direction of P3HT domains. The reason of the above variations is tentatively assigned to hydrophobic forces acting on P3HT chains being in the polar environment, i.e., PMMA matrix, which forces P3HT aggregates to conform a specific arrangement inside the matrix. Such a process is most effective for smaller P3HT particles since the most influence is rendered onto the molecules being at the interface of P3HT-PMMA. Additional evidence that supports the above suggestion is the fact that the ratio of the first absorption maximum in respect to the sidebands decreases with time, which implies that the equilibrium between the different crystalline forms of P3HT in PMMA matrix evolves, namely, the form I’ gradually converts to the more thermodynamically stable form II (Fig. 10). Such a result reflects slow relaxation processes in PMMA matrix itself that acts on P3HT domains and thus renders a delayed effect which is more pronounced in samples with smaller P3HT particles.

Electronic absorption spectra of as-prepared samples (solid curves ) and the same samples 2 weeks later (dotted curves ) of P3HT-PMMA (1:50 weight ratio, red lines ) and P3HT-PMMA (1:4 weight ratio, blue lines )
结论
An increasing QY of PL which has been found in P3HT particles embedded into PMMA matrix is an unusual phenomenon since it takes place when the polymer molecules are still aggregated and where a strong exciton quenching should be normally observed. The increasing QY is assigned due to the two factors. The minor factor is the changing dielectric constant which facilitates a modest increase in QY by about 14%. The major factor is due to rearrangement of the polymer chains themselves. Better chain ordering in P3HT domains embedded into the PMMA matrix has been unequivocally proved by spectroscopy methods and calculation of the exciton bandwidth as well. The reason of the structural changes is tentatively assigned to hydrophobic forces acting on P3HT chains being in polar environment, i.e., PMMA matrix, which forces P3HT aggregates to conform a specific arrangement inside the matrix. Such a process is most effective for smaller P3HT particles since the most influence is rendered onto the molecules being at the interface of P3HT-PMMA. Particularly, it can be concluded that the composite P3HT-PMMA samples contain P3HT crystals of two forms, i.e., I’ and II, in which the interchain stacking distance varies from 0.42 to 0.44 nm. In form I’, intramolecular torsional disorder is reduced and most probably it promotes the increasing coherent domain size in the π − π stacking direction of P3HT domains, respectively. This is accompanied by the increasing first absorption maximum in respect to sidebands in spectra of composite P3HT films and by narrowing free exciton bandwidth, respectively. It is interesting to note that the narrowing exciton bandwidth is a factor which is responsible for increasing QY of PL emission in semiconductor nanoparticles as compared to the bulk crystals possessing wide energetic bands [51]. Narrow bands reduce smearing effect upon electronic transitions, thus facilitating more electrons falling to the same energy level. Thus, the observed enhanced QY of emission of P3HT particles can be interpreted in terms of the changing intermolecular packing and reduced intramolecular torsional disorder along with narrowing exciton bandwidth.
纳米材料