

hGC33 修饰和索拉非尼负载的纳米颗粒通过抑制 Wnt 信号通路具有协同抗肝癌作用
摘要
肿瘤特异性抑制剂的递送是癌症治疗中的一个挑战。抗体修饰的纳米粒子可以将其负载的药物递送至过度表达特定肿瘤相关抗原的肿瘤细胞。在这里,我们构建了载有索拉非尼的聚乙二醇-b-PLGA 聚合物纳米颗粒,该纳米颗粒用抗体 hGC33 修饰,以抵抗 glypican-3(GPC3 +),一种在肝细胞癌中过表达的膜蛋白。我们发现 hGC33 修饰的 NPs (hGC33-SFB-NP) 靶向 GPC3 + 肝细胞癌 (HCC) 细胞通过与 HCC 细胞表面的 GPC3 特异性结合,抑制 Wnt 诱导的信号转导,并通过下调细胞周期蛋白 D1 的表达来抑制 G0/1 期的 HCC 细胞,从而通过抑制上皮-间质转化。 hGC33-SFB-NP 通过分别与 GPC3 分子协同抑制 Ras/Raf/MAPK 通路和 Wnt 通路来抑制 HCC 细胞的迁移、循环进程和增殖。 hGC33-SFB-NP在体内抑制肝癌的生长,提高荷瘤小鼠的存活率。我们得出结论,hGC33 增加了 SFB-NP 对 HCC 细胞的靶向。 hGC33-SFB-NP通过阻断Wnt通路和Ras/Raf/MAPK通路协同抑制HCC的进展。
介绍
Glypican3 (GPC3) 是一种硫酸乙酰肝素蛋白多糖,通过涉及甘油磷脂酰肌醇锚的机制在细胞表面表达 [1]。尽管 GPC3 在发育过程中在多种组织中表达,但其表达在大多数成人组织中至少部分受到启动子区域 DNA 甲基化的抑制 [2]。然而,GPC3 蛋白在约 70% 的肝细胞癌 (HCC) 患者中过度表达 [3, 4] 并通过与 Wnt 配体相互作用刺激经典的 Wnt 信号转导 [5],从而促进 Wnt/卷曲结合以促进 HCC 生长 [6] .经典 Wnt 信号通路的激活是与 HCC 恶性转化相关的最常见事件之一 [7, 8]。基于 GPC3 增加 Wnt 信号的能力,我们假设 GPC3 的过表达通过刺激经典的 Wnt 通路促进 HCC 生长。
识别 GPC3 (524–563) (hGC33) C 端表位的治疗性单克隆抗体,可识别 GPC3 (524–563) 的 C 端表位,抑制 HepG2 和 Huh- 皮下异种移植物中的肿瘤生长7 小鼠 [9, 10]。 hGC33已通过互补决定区移植人源化,其抗癌作用与hGC33对HepG2异种移植一样有效,hGC33已用于临床试验[11]。这些结果表明hGC33具有重要的抗肿瘤活性,抗GPC3治疗将通过阻断Wnt和/或其他信号通路直接抑制HCC细胞的增殖和/或存活。
然而,由于 HCC 的复杂发病机制,单独阻断 GPC3 的功效是有限的 [12]。因此,探索HCC发病机制的详细机制,并为HCC的诊断和预后寻找有前景的生物标志物,可能有助于提供有效的治疗靶点,改善患者的预后。已提出抗 GPC3 抗体可增加 HCC 对化疗药物的敏感性 [13]。最近评估了 hGC33 与标准化疗药物联合使用的抗肿瘤活性 [14]。索拉非尼是一种新型的口服靶向 HCC 药物,可通过阻断 RAF/MEK/ERK 介导的细胞信号通路直接抑制肿瘤细胞的增殖,从而抑制肿瘤生长 [15, 16]。然而,索拉非尼在体内的非靶向分布和 HCC 中异常激活的 Wnt 信号限制了药物的有效性并增加了其副作用 [14,15,16]。 Wnt与Frizzled家族受体结合,激活细胞内信号转导,调节细胞增殖、凋亡和细胞迁移,并在多种肿瘤如HCC中引起耐药[17,18,19]。
在 HepG2 异种移植模型中,hGC33 和索拉非尼的组合比单独使用索拉非尼更有效地抑制肿瘤生长[15],并且聚合物纳米载体的药物递送在癌症治疗中备受关注。载抗癌药物的纳米载体可以防止体内非特异性分布和非特异性降解,提高药物生物利用度和抗肿瘤靶向性,简化药代动力学和治疗的评价[15, 16]。在各种基于聚合物的纳米粒子中,基于聚(乳酸乙醇酸)(PLGA)的制剂被认为是理想和安全的药物载体 [17, 18]。在这方面,聚(乙二醇)-b -聚(d,l-丙交酯-共-乙交酯)(PEG-b -PLGA) 是基于聚乙二醇和 PLGA 共聚物,水解后安全无毒,已获得美国食品和药物管理局的批准 [19,20,21,22]。因此,PEG-b纳米载体静脉注射 -PLGA 共聚物是实现靶向递送和提高功效的有前景的策略。此外,在HCC细胞膜上应用针对GPC3分子的特异性抗体hGC33,不仅可以提高纳米药物靶向的体外和体内递送[18, 19],还可以阻断Wnt和/或其他与GPC3相关的信号通路,抑制癌细胞的增殖和/或存活,并可能实现协同抗肿瘤活性。
在本研究中,我们探讨了 hGC33 抗体修饰的共聚物 PEG-b -PLGA纳米粒可促进索拉非尼(hGC33-SFB-NP)在体内外递送至HCC,并通过HCC活性靶向改变药物药代动力学提高HCC治疗效率。根据粒径、zeta电位、颗粒形貌、药物包封率、载药量和体外药物释放,对靶向NP进行了综合表征。体外靶向能力的特点是细胞摄取 HepG2 肝癌细胞。通过比较索拉非尼和SFB-NP,评估hGC33-SFB-NP对HCC的生物分布和协同治疗作用。我们的结果表明 hGC33-SFB-NP 可以靶向 GPC3 + 肝细胞癌。可通过抑制Wnt和Ras/Raf/MAPK信号通路抑制细胞周期进程、细胞增殖和肿瘤侵袭,协同抑制肝癌进展。
材料和方法
材料
hGC33 抗体,3-(4,5-二甲基噻唑-2-基)-2,5-二苯基溴化四唑 (MTT),4',6-二脒基-2-苯基吲哚,二盐酸盐 (DAPI),5,5',6 ,6'-四氯-1,1',3,3'-四乙基-苯并咪唑基碘化碳菁 (JC-1) 和二甲基亚砜 (DMSO) 购自 Sigma-Aldrich, Inc. (St. Louis, MO)。 1-(3-二甲氨基丙基)-3-乙基-碳二亚胺盐酸盐和N -羟基琥珀酰亚胺购自启云生物技术公司(中国广州)。二辛可宁酸(BCA)蛋白定量试剂盒、香豆素-6、Annexin V-FITC/PI细胞凋亡检测试剂盒购自碧云天生物(中国上海)。 PEG-b -PLGA-马来酰亚胺二嵌段共聚物(mal-PEG-b -PLGA; 25,000–30,000 Da,PLGA,LA:GA,w/w; PEG, 13–15%) 购自 Polyscitech (West Lafayette, IN, USA)。 PI3K抗体试剂盒(9655#)、p-Akt抗体试剂盒(9916#)、mTOR抗体试剂盒(9964#)、Bcl-2家族抗体试剂盒(9942#)、凋亡抗体试剂盒(9915#)、山羊二抗兔和抗小鼠抗体购自 Cell Signal Technology (Danvers, MA, USA);细胞周期蛋白 B1 和细胞周期蛋白依赖性激酶购自 Abcam Biological Technology (USA)。磷酸化 Rb、细胞周期蛋白 D1、检查点激酶 1 (CHK1)、P53、磷酸化乳腺癌易感基因 1 (p-BRCA1)、RAD51、细胞色素 C 和基质金属蛋白酶(MMP2 和 MMP9)的抗体购自 Abcam(美国) .所有其他分析级化学品、试剂和溶剂均从标准供应商处获得,无需进一步纯化即可使用。
细胞和动物
HCC 细胞系 HepG2(从美国典型培养物保藏中心(Manassas,VA,USA)获得)在补充有 10% FBS(HyClone,Logan,UT,USA)和 80 U/ml 青霉素和 80 μg/ml 链霉素,在 5% CO2 的潮湿环境中,37°C。BALB/C 裸鼠,体重 20-22 克(5-6 周)由南京骏科生物技术有限公司提供. (中国). BALB/C裸鼠在SPF室饲养,所有动物护理和治疗均按照安徽科技大学动物护理要求进行,所有实验方案均已通过动物实验审查和批准安徽科技大学伦理委员会(批准文号:2019dw013)。
NP 的准备
制备 NP,mal-PEG-b -PLGA 和 SFB 或香豆素-6 称重并溶解在有机相中(3:2 v/v 二氯甲烷/丙酮)。将溶液逐滴加入到聚乙烯醇 (PVA) 溶液 (5% w/v) 中,同时连续涡旋。在冰上用探头超声仪(输出功率为 550 W,8 次)间歇性地对混合物进行超声处理,以产生油水乳液。在磁力搅拌下将乳液加入到 PVA (1% w/v) 溶液中。通过以 8000 rpm 离心 30 分钟收集 SFB 和香豆素-6 NP,并在 Milli-Q 水中洗涤 3 次。
为了通过马来酰亚胺与抗体 hGC33 中的游离巯基残基反应形成的硫醚键生成 hGC33-NP,将 hGC33 抗体与马来酰亚胺官能化的 NP 以 5:1 (hGC33:mal-PEG-b) 的摩尔比混合 -PLGA) 并在 4°C 下持续搅拌孵育 16 小时。 hGC33 通过抗体 hGC33 上的巯基与 PEG 链的马来酰亚胺基团的反应与 NP 偶联。通过Sepharose CL-4B柱去除未结合的抗体。使用 BCA 试剂盒 (Thermo Fisher Scientific, Waltham, MA, USA) 确认了有效的蛋白质缀合。
纳米粒子 (NP) 的表征
纳米颗粒的形态、粒径、封装效率 (EE) 和稳定性
用透射电子显微镜(TEM,H-600;Hitachi,Tokyo,Japan)评估纳米颗粒的形态。使用溴化钾通过 FTIR 分光光度计(Thermo Nicolet,麦迪逊,威斯康星州,美国)记录不含药物的 hGC33-NPs 和不含药物的 NPs。使用 Malvern Zetasizer ZEN3600 Nano ZS(Malvern Instruments,Malvern,UK)在 20°C 下表征 NP 的平均粒径和 zeta 电位。通过超滤评估药物包封率(EE)和载药量(LC)效率。将样品装入超滤装置(100 kMWCO;Sartorius,Göettingen,Germany)并在 4°C 下以 8000 rpm 离心 25 分钟以去除游离药物。将每个样品的相同体积溶解在乙腈中以确认药物的总量。浓度用高效液相色谱法测定。吸收波长为 266 nm。以下公式用于计算NPs的药物EE(%):(包裹的药物重量/药物总重量) × 100%。 LC (%) 计算为(包封药物的重量/NPs 的重量) × 100%。为了了解 NPs 在室温下的稳定性,通过动态光散射 (DLS) 在预定时间点(0.5、1、2、4、8、12 小时、16 小时、20 小时和 24 h) 在 25°C。
体外药物释放和细胞摄取测定
使用截留分子量为 10 kDa 的透析袋研究来自 NPs 的药物。简而言之,将 1 mL NP 装入透析袋(MWCO 8,000–10,000 Da;Spectrum Labs Inc., CA, USA)。将透析袋浸入 PBS 中并在 25°C 下用磁力搅拌器搅拌。在 100 mL 的 0.2 M 磷酸盐缓冲盐水(PBS;pH =7.4)中测量 7 天的 NP 的药物释放曲线。样品中药物浓度采用高效液相色谱法测定。在随后的研究中,使用与 hGC33-SFB-NP 具有相同粒径的 hGC33-香豆素 6-NP 来评估 hGC33-SFB-NP 的靶向性。 HepG2 (GPC3 + ) 和 Li-7 (GPC3 − ) 分别与 hGC33-香豆素 6-NP 在 37°C 和 5% CO2 中孵育 0.5、2 或 4 小时。将共培养的细胞洗涤并用 4% 甲醛固定 10 分钟;细胞核用 5 μg/mL Hoechst 33,342 染色 15 分钟以定位细胞内 NP。用共聚焦显微镜(Olympus FV1000;Olympus Corporation,Tokyo,Japan)分析用于分析细胞内纳米颗粒图像的共聚焦显微镜。
体外细胞效应
使用 MTT 测定法测定游离 hGC33(Ab)、游离 SFB、hGC33-null-NP 或 hGC33-SFB-NP 的细胞毒性。 HepG2(GPC3 + ) 细胞和 Li-7 (GPC3 − ) 对数期的细胞以每孔 4000 个细胞的密度接种到 96 孔板中,然后在 37°C 下在 5% CO2 中孵育 48 小时。细胞在 37°C 和 5% CO2 中用 hGC33、游离 SFB、hGC33-null-NP 或 hGC33-SFB-NP 处理 48 小时。共培养一定时间后,如[20]所述通过MTT测定法测定抗细胞增殖活性。 490 nm处测定各孔吸光度,用SPSS 17.0计算最大抑菌浓度值的一半(IC 50)。
细胞侵袭能力的测量
将处于对数生长期的细胞以5 × 10 4 的密度接种于6孔板 细胞/孔,用吸头刮擦,并更换为无血清培养基。对照组和实验组在 0 小时、24 小时和 48 小时记录伤口愈合情况。同时,将相同数量的细胞接种到 Transwell 小室中,并用游离 hGC33、游离 SFB、hGC33-null-NP 或 hGC33-SFB-NP 处理。将五百微升 10% FBS 培养基加入下室。孵育24小时后,取出Transwell小室,4%多聚甲醛固定细胞,0.1%结晶紫染色。计算伤口愈合的大小和迁移细胞的数量来评价迁移能力。
细胞周期测定
在 6 孔板中孵育过夜后,将细胞用游离 hGC33 (Ab)、游离 SFB、hGC33-null-NP 或 hGC33-SFB-NP 处理 24 小时,然后收集并用乙醇固定。碘化丙啶染色后进行流式细胞术,用modifit 3.0(Verity Software House, Topsham, ME)分析细胞周期。
蛋白质印迹
为了评估信号通路的激活状态和靶分子的表达,将细胞在 6 孔板中孵育过夜,并施加游离 hGC33、游离 SFB、hGC33-null-NP 或 hGC33-SFB-NP 24 小时.收集各处理组细胞,提取蛋白质并测定。使用 BCA 蛋白质试剂盒(Biosharp,合肥,中国)测量和校准蛋白质浓度。样品蛋白质经十二烷基硫酸盐聚丙烯酰胺凝胶电泳分离,转移至PVDF膜,无脂牛奶封口。第一抗体(1:1000 稀释)在 4°C 下孵育过夜,第二抗体(1:2000)在室温下孵育 1 小时。用ECL底物(Thermo Fisher Scientific Waltham,MA,USA)观察条带,凝胶图像分析系统显示图像,以β-actin为对照。
体内抗肿瘤活性
测定了游离 hGC33、游离 SFB、hGC33-null-NP、SFB-NP 和 hGC33-SFB-NP 对 HCC 体内生长的抑制作用。根据安徽工业大学伦理委员会关于动物健康的规定和指南,所有实验均在 BALB/c 小鼠的笼子中进行,温度控制室(23 ± 2°C),12 h/12 h 光照/黑暗循环。含有 5 × 10 6 的 50 μl 悬浮液 将活的 HepG2 细胞皮下注射到 5 周龄雌性 BALB/c 小鼠(20-22 克)的右腹部。当肿瘤体积达到50mm左右 3 ,将小鼠随机分为6组(每组10只)。生理盐水对照 NS(200 μL PBS 中的 200 μg/kg null NP)、hGC33-null-NP(200 μL PBS 中的 hGC33-null-NP,相当于 hgc33 =100 μg/kg/次)、游离 hGC33(hGC33 in 200 μL PBS,100 μg/kg/次)、游离 SFB(SFB 剂量:8 mg/kg/次)、SFB-NP(SFB 剂量:8 mg/kg/次)和 hGC33-SFB-NP(相当于SFB =8mg/kg/次,hGC33 =100μg/kg/次)每2天通过尾静脉注射10次。每四天测量一次小鼠的体重和肿瘤大小。肿瘤体积的计算公式为体积 =0.5 × L × W 2 ,其中 L 和 W 分别代表肿瘤的长度和宽度。给药4周后,用乙醚麻醉动物,测量肿瘤大小和重量。此外,切除肿瘤、心、肝、肾、肺、脾,4%多聚甲醛溶液固定,石蜡包埋,切片,苏木精伊红染色,数码显微镜观察组织学变化。
统计分析
数据表示为平均值 ± 标准偏差(SD),并通过使用 SPSS 18.0 的方差分析进行评估。使用双尾学生 t 检验进行成对统计比较。 P 的差异被认为具有统计学意义 <0.05.
结果
NPs 的表征和药物释放 体外
由于纳米颗粒的直径和表面特性会影响细胞吸收、药物释放和纳米颗粒在体内的分布,我们用相应的参数表征了制备的纳米颗粒。 SFB-NP、hGC33-null-NP和hGC33-SFB-NP聚合物的形貌、粒径和粒径分布总结在图1中。hGC33-SFB-NP的形貌通过透射电子显微镜观察到(图1) . 1a) 显示具有模糊边缘的刚性核,这表明 hGC33 存在于 NP 表面。 SFB NP、hGC33-null-NP 和 hGC33-SFB-NP 的粒径范围为 100 至 150 nm,具有典型的单峰粒径分布。 hGC33-SFB-NP 的平均直径(120.2 ± 10.2 nm)略大于 hGC33-null-NP、SFB-NP 和 null NP(图 1a,表 1)。 hGC33-SFB-NP和hGC33-null-NP表面存在单面球形不透明薄膜,表明NPs表面存在抗体hGC33。 hGC33-SFB-NP 和 hGC33-null-NP 的尺寸增加证实了 hGC33 膜的存在。 hGC33-SFB-NP的合成通过 1 确认 NP 制备前的 H-NMR(图 1b)。 5.2 ppm 和 1.58 ppm 处的峰归属于乳酸的 -CH3 质子; 4.8 ppm 处的峰归属于乙醇酸的 -OCH2-; 3.6-3.8 ppm 处的峰被指定为 PEG 重复单元的 -CH2CH2O- 质子。 PEG-b 的表面化学 -PLGA 和 Ab-PEG-b -PLGA NPs 也通过 FTIR 光谱进行了研究(图 2c)。在 PEG-b 的光谱中 -PLGA 聚合物,约 1750 cm -1 处的强带 源于 PLGA 链中羰基(C =O)的拉伸。 2880 cm −1 处的波段 是由于 PEG 链中 a-CH 基团的拉伸所致。同时,在 2520 cm −1 处出现一个峰 我们将其归因于 hGC33 抗体的 -SH 拉伸峰。 1 H NMR和FTIR结果表明抗体接枝在PEG-b的骨架上 -PLGA聚合物。
<图片>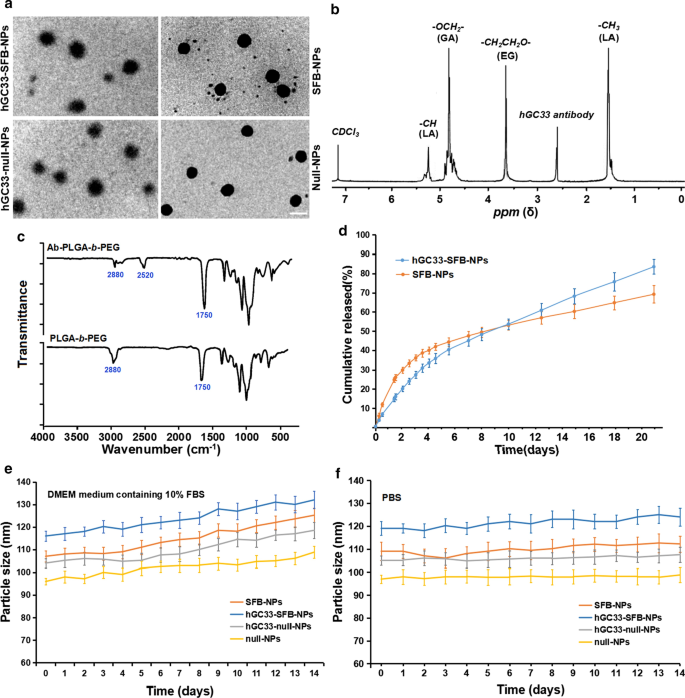
NP 的表征。 一 NPs 的 TEM 表征,比例尺表示 100 nM; b 1 合成hGC33-PLGA-b的H NMR谱 -CDCl3中的PEG; c hGC33-PEG-b的FTIR光谱 -PLGA 和 PEG-b - PLGA; d SFB-NP 和 hGC33-SFB-NP 在 PBS (pH =7.4) 中 37°C 的累积释放曲线; e 在含有 10% FBS 的 DMEM 培养基中孵育 14 天后 NPs 的大小变化; f 在 PBS 中孵育 14 天后 NPs 的大小变化。 SFB,索拉非尼; TEM,透射电子显微镜;纳米粒子,纳米粒子; 1 核磁共振, 1 H-核磁共振波谱; FTIR、傅里叶变换红外光谱
<图片>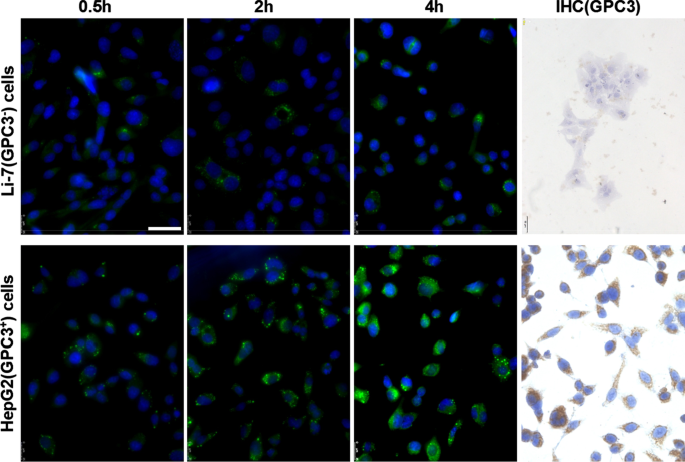
Li-7 和 HepG2 细胞中 GPC3 的表达和 hGC33-香豆素6-NP 的摄取。接种在培养板中的细胞用 PBS 洗涤,并与 DMEM 中的 100 μg/ml hGC33-香豆素 6-NP 孵育 2、4 和 8 小时。细胞核用 DAPI 染色,细胞固定并通过共聚焦激光扫描显微镜检测。 GPC3 在 Li-7 细胞中未检测到,但通过免疫细胞化学检测在 HepG2 细胞中高度表达。比例尺表示 50 μM
有趣的是,hGC33-SFB-NP 和 SFB-NP 纳米颗粒在含有 10% FBS(pH =7.4)的 DMEM 培养基中的药物释放迅速,直到大约 4 天,然后药物释放相对缓慢和稳定; hGC33-SFB-NP 和 SFB-NP 在 20 天内的累积 SFB 释放量分别约为 77% 和 65%(图 1d)。差异可能是由于 PEG-b 表面的亲水性分子 -PLGA 基质,它可以通过增加水合作用加速纳米颗粒的降解,从而促进水解。为了确定制备的 NPs 的稳定性,将 hGC33-SFB-NP、hGC33-null NP、null-NP 和 SFB-NP 置于含有 10% FBS(pH =7.4)和 PBS(pH =7.4)的 DMEM 培养基中)。各种 NP 的大小保持稳定超过 2 周。 SFB 以持续稳定的方式从 hGC33-SFB-NP 释放 14 天,但与 10% FBS 相比,DMEM 培养基中的颗粒大小发生了适度变化(图 1e、f)。 hGC33-SFB-NP的稳定性适合于具有持续生物学作用的SFB。
较大的 zeta 电位会引起 NP 之间强烈的静电排斥相互作用,并保持 NP 分散体系的稳定性 [23, 24]。如表1所示,hGC33-SFB-NP、hGC33-null-NP和SFB-NP的zeta电位分别为 − 18.2 ± 2.2 mv、 − 18.5 ± 1.8 mv和 -9 15 mv.由抗体 hGC33 糖蛋白中的醛基、羧基和磷酸基产生的负电荷引起。带负电荷的 NPs 可以导致 NP 悬浮的高稳定性。此外,由于细胞表面在正常生理环境中带负电荷,所制备的纳米粒子排斥低电荷细胞,对组织和细胞的毒性较小。此外,null-NP 和 SFB-NP 的尺寸分布(多分散指数 [PDI] 分别为 0.18 和 0.19)比 hGC33-null-NP (PDI =0.21) 和 hGC33-SFB-略小但不显着小NP(PDI =0.23)。因此,纳米粒子的尺寸具有良好的均匀性。根据SFB包封率和负载量的结果,NP的粒径、粒径分布和zeta电位见表1。通过超速离心去除未结合的 GPC3 抗体 hGC33,并评估 hGC33 抗体与 NPs 的结合效率。 BCA蛋白分析表明hGC33抗体与NPs的结合率为79.5% ± 2.9%。
hGC33-Coumarin 6-NP 有效靶向 GPC3 + HCC 细胞系 HepG2 细胞
为了确定纳米颗粒的hGC33抗体是否仍然具有特异性靶向GPC3的能力,我们使用了GPC3 + HepG2 和 GPC3 − Li-7 细胞作为靶细胞,hGC33-香豆素 6-NP 作为示踪剂纳米颗粒,并将不同的细胞孵育 2 小时、4 小时和 8 小时。用PBS洗涤细胞3次并与DAPI反应以染色细胞核。将细胞固定并用 Leica 荧光显微镜(DMi8,德国)拍照。发现HepG2细胞的绿色荧光在相同的孵育时间明显高于Li-7细胞(图2),说明hGC33-香豆素6-NP进入HepG2细胞的量明显高于Li-7细胞在 Li-7 电池中。结果证明 hGC33-Coumarin6-NP 上的 hGC33 抗体仍然具有靶向 GPC3 并介导纳米颗粒内化的能力。 GPC3 在 HepG2 细胞和 Li-7 细胞中的表达通过间接荧光和细胞化学染色检测。结果显示HepG2细胞过表达GPC3,而Li-7细胞不表达GPC3(图2)。
hGC33-Null-NP 抑制 HepG2 细胞增殖
为了确定 hGC33 修饰的 NP(hGC33-null-NP)是否可以抑制 HCC 的生长,我们测量了 GPC3 阳性 HCC 细胞系 HepG2 和 GPC3 阴性细胞系 Li-7 的细胞生长抑制。我们发现 hGC33-null-NP 和 hGC33 在处理 24 小时后均抑制 GPC3 阳性 HCC 细胞系 HepG2 的生长,但 hGC33 对 HepG2 细胞的抑制作用比 hGC33-null-NP 更显着(图 3a);相比之下,hGC33-null-NP 和 hGC33 不影响 GPC3 阴性 Li-7 细胞系的生长(图 3b)。 hGC33-null-NP(相当于 0.1 μ m hgc33)和 hGC33(0.1 μ m)对 HepG2 细胞增殖的代表性时间过程如图 3c 所示。由于 hGC33 是识别 GPC3 的 C 端肽,因此 hGC33 在抑制 HepG2 方面优于 hGC33-null-NP。结果表明hGC33-null-NP中的hGC33分子可能具有空间阻断作用,可能影响hGC33与表位的结合能力。
<图片>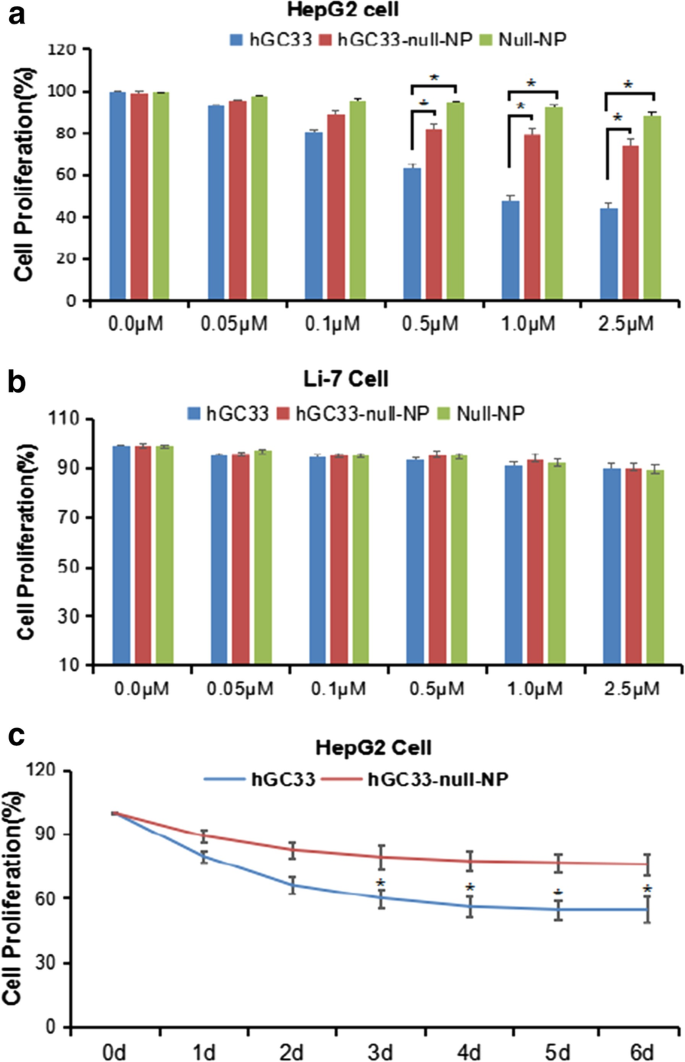
HepG2 和 Li-7 细胞的增殖受到 hGC33、null-NP 和 hGC33-null-NP 的抑制。 一 对用hGC33、null-NP或hGC33-null-NP处理的GPC3阳性HepG2细胞进行细胞增殖测试; b 对用 hGC33、null-NP 和 hGC33-null-NP 处理的 GPC3 阴性 Li-7 细胞进行细胞增殖测试。将细胞与 0–2.5 μM hGC33、null-NP 或 hGC33-null-NP 孵育 1 天。用MTT方法测量细胞的增殖并标准化为未处理的细胞。所有值代表平均值 ± SD。与未经抗体处理的对照组(0 μM)相比,*P <0.01; c 在 hGC33 处理中 HepG2 对 hGC33、null-NP 和 hGC33-null-NP 反应的代表性结果 (1.0 μM)
hGC33-Null-NP 抑制 HepG2 细胞的细胞周期
为了了解在 hGC33-null-NP 纳米颗粒上修饰的 hGC33 抗体的分子活性机制,我们研究了用 hGC33-null-NP 处理的 HepG2 的细胞周期进程。在 HepG2 细胞系中,hGC33-null-NP 和 hGC33 显着增加了 G1 期细胞的比例(图 4a、b),表明 hGC33-null-NP 和 hGC33 都可以诱导 G1 期细胞周期停滞。此外,hGC33-null-NP和hGC33可显着下调HepG2细胞中cyclinD1的表达(图4c、d)。
<图片>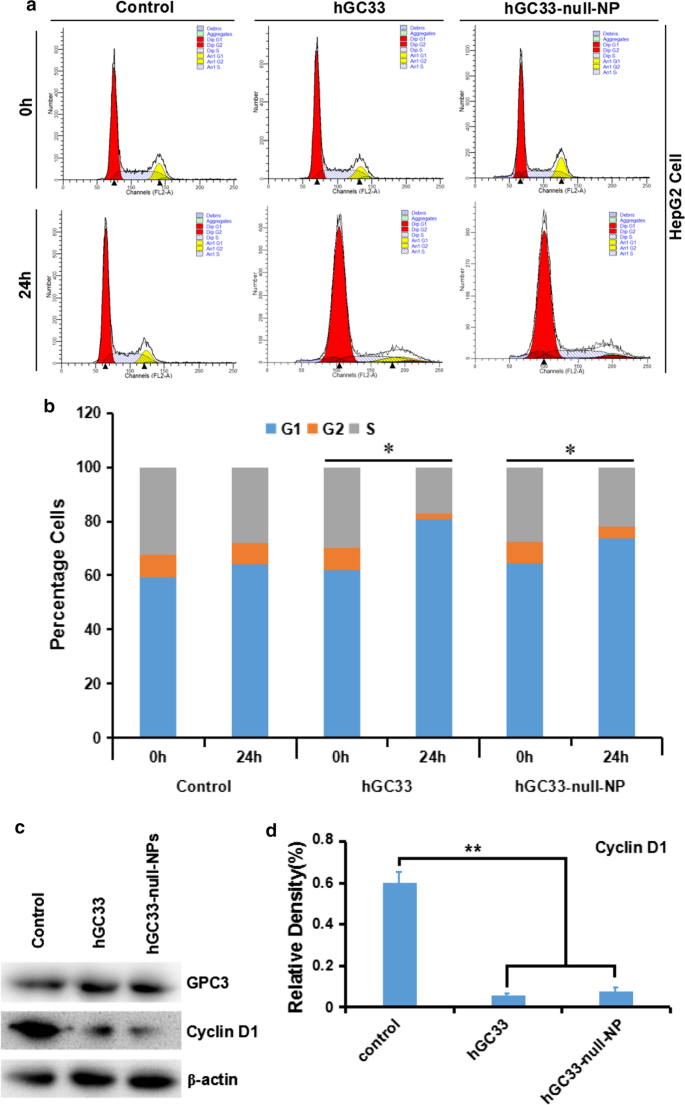
hGC33-null-NP 和 hGC33 诱导细胞周期停滞在 G1 期并抑制 HepG2 细胞中的细胞周期蛋白 D1 表达。 一 用 hGC33-null-NP 和 hGC33 处理的各组的代表性细胞周期图。 b 用 hGC33-null-NP 和 hGC33 处理的各组的细胞周期分析。将 HCC 细胞与 0.5 μm hGC33 或 hGC33-null-NP(以 hGC33 的摩尔浓度作为参考)一起温育。 *P <0.05,将 hGC33-null-NP 或 hGC33 细胞的 G1 期与 0 h 细胞的 G1 期进行比较。 c , d hGC33-null-NP或hGC33处理后,与对照组相比,HepG2细胞中cyclinD1显着下调
HepG2、Huh-7 和 Li-7 细胞中的 Wnt 激活
为了了解 HCC 细胞中经典 Wnt/β-Catenin 信号的激活,我们首先检测了各种 HCC 细胞系中 Wnt 配体和受体卷曲蛋白(卷曲、FZD 或 Frz)的表达:HepG2 (GPC3 ++ ), Huh-7 (GPC + ) 和 Li-7 (GPC3 − )。结果表明,转导β-Catenin通路的Wnt3a和FZD受体在所有细胞系中均有表达,尤其是在HepG2和Huh-7细胞中(图5)。
<图片>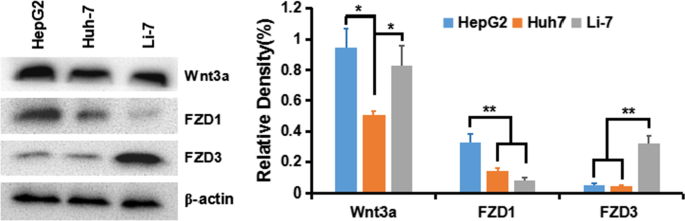
Wnt3a、FZD1、FZD3在肝癌细胞系中的表达
hGC33-Null-NP 抑制 Wnt3a 诱导的 Wnt 信号转导依赖性细胞增殖
有研究表明,GPC3的胞外部分可能是Wnt的共同受体,促进Wnt/β-Catenin信号的激活和转导。因此,当 HepG2 (GPC3 + ), Huh-7 (GPC3 + ) 和 Li-7 (GPC3 − ) cells were co-incubated with hGC33 and hGC33-null-NP, the activation of Wnt/β-catenin signal was blocked by hGC33 and hGC33-null-NP, and proliferation of HepG2 and Huh-7, but not Li-7, in Wnt3a-conditioned medium was inhibited. That proliferation of Li-7 cells most likely is due to the absence of GPC3 on the surface of Li-7 cells (Fig. 6). Our results indicate hGC33 and hGC33-null-NP nanoparticles specifically bind to GPC3 molecules on cell membrane. hGC33 and hGC33-null-NP partially neutralize the mitogenic activity of Wnt3a and inhibit the Wnt/β-catenin signaling pathway. However, the inhibition of proliferation by hGC33-null-NP nanoparticles was less than that of hGC33. Perhaps the spatial structure of the nanoparticles interferes with hGC33-null-NP and limits the function of the hGC33 molecule on the nanoparticles so they cannot completely block the interaction between GPC3 and Wnt3a.

GPC3-activated Wnt signal transduction in HCC. Fifty percent Wnt3a DMEM medium was added with anti-wnt3a antibody, hGC33, or hGC33-null-NP. HepG2 (GPC3 ++ ), Huh-7 (GPC3 + ) and Li-7 (GPC3 − ) cells were co-incubated for 48 h, and cell proliferation was measured by MTT assay. The data were expressed as mean ± SD (*P < 0.01)
hGC33-Null-NP Inhibits Wnt3a-Induced Signal Transduction in HepG2 and Huh-7 Cells
To understand the effect of hGC33-null-NP on Wnt/β-catenin signaling in HCC cells, we extracted the proteins of HepG2 and Huh-7 cells treated with hGC33 or hGC33-null-NP. The results of western blot showed that after hGC33-null-NP treatment, the levels of pYAP and pβ-catenin were increased, but the levels of YAP and β-catenin were decreased. Furthermore, the levels of cyclinD1, CD44, VEGF, and c-MYC in the hGC33-null-NP group were lower than those in the control group, but the level was less than with hGC33 treatment. Similar effects were observed in HepG2 and Huh-7 cells, as shown in Fig. 7.

Inhibition of Wnt3a-induced β-catenin signaling by hGC33-null-NP or hGC33. 一 Compared with the control group, Wnt/β-catenin signaling pathway in HepG2 and Huh-7 cells treated with hGC33-null-NP or hGC33 was inhibited, and the levels of β-catenin and YAP were decreased, while the increased phosphorylated β-catenin and phosphorylated YAP molecules were unstable, and degraded in cytoplasm. The decreased β-catenin was difficult to maintain in the nucleus and drive the expression of CyclinD1, CD44, VEGF, and c-MYC, which resulted in the decrease of cyclinD1, CD44, VEGF, and c-myc protein levels. b The mechanism pattern of Wnt/β-catenin signaling pathway inhibited by hGC33-null-NP or hGC33
hGC33-SFB-NP or hGC33 Attenuates HCC Cell Migration by Inhibiting Epithelial Mesenchymal Transition (EMT)
HCC is one of the deadliest cancers in the world, and its incidence is steadily increasing. Sorafenib is the only approved standard treatment for patients with advanced HCC, as it has been shown to improve the survival rate of these patients. However, clinical and preclinical observations suggest that sorafenib therapy has limited efficacy due to the rapid development of drug resistance. Therefore, elucidating the mechanism of escape resistance to sorafenib is a major emphasis in HCC research. In recent years, more and more attention has been paid to the role of epithelial mesenchymal transition (EMT) in the progress of HCC and the development of drug resistance. EMT refers to the transformation of epithelial to mesenchymal cells, which endows cells with the ability metastasize and invade, including acquisition of stem cell characteristics, reducing apoptosis and aging, promoting immunosuppression, and participating in the occurrence and development of cancer. The loss of E-cadherin expression is considered a key step in the carcinogenesis and EMT. EMT is a developmental multi-step molecular and cellular reprogramming process that cancer cells use to achieve aggression. This is mainly through down regulating the expression of E-cadherin, keratin, mucin, ZO-1 (tight junction protein); up regulating the expression of vimentin, alpha-smooth muscle actin (α-SMA), FN fibronectin, MMPs (degradation matrix), N-cadherin, snail, slug, twist, Rho, TGF-β, FGF, type I collagen, and type II collagen to achieve the invasion, metastasis, and anti-apoptosis of EMT characteristic tumor. The changes of these protein expressions mainly involve the activation of Wnt/β-catenin and Ras/Raf/MAPK signaling pathways.
Our experiments have shown that hGC33 antibody on the surface of NP vector can inhibit Wnt3a-induced β-catenin signal transduction, and then down regulate the expression of β—catenin, CD44, vascular endothelial growth factor (VEGF), cyclin D1, and c-MYC. Furthermore, hGC33-SFB-NP inhibits the activation of Ras/Raf/MAPK signal pathway and inhibits proliferation and apoptosis of HCC cells. hGC33 and SFB have a synergistic effect, inhibiting EMT and decreasing the migration of HCC cells (Fig. 8).

Effect of hGC33-SFB-NP on EMT inhibition. 一 Compared with the control group, the hGC33-SFB-NP treatment group had less cell migration. Photographs were taken under an optical microscope (magnification, × 200). The error value represents the standard deviation of three independent experiments. *Compared with the control group, p <0.01。 b Compared with the control group, the EMT-related proteins snail, vimentin, and MMP-2 in HCCs treated with hGC33-SFB-NP decreased, whereas E-cadherin increased. c Molecular mechanism of EMT. EMT, epithelial–mesenchymal transition; MMP-2, matrix metalloproteinase-2; SFB, sorafenib; NP, nanoparticle
hGC33-SFB-NP Inhibits the Growth of Hepatocellular Carcinoma In Vivo and Improves the Survival Rate of Tumor-Bearing Mice
To evaluate the anti-tumor activity of hGC33-SFB-NP in vivo, HepG2 and Huh-7 cells were inoculated subcutaneously into the right abdomen and dorsal side of female BALB/c nude mice, respectively. When the tumor xenograft growth reached about 30 mm 3 , the mice were randomly divided into groups to further evaluate the inhibition of each group (hGC33-SFB-NP, hGC33-null-NP, SFB-NP, free hGC33, free SFB, and control group) HCC effect (n = 5 per group). It can be seen from Fig. 9a, b that hGC33-SFB-NP significantly slowed tumor growth in mice compared with the PBS control and other treatments. Compared with the PBS control, hGC33-null-NP, SFB-NP, free hGC33, and free SFB also had some inhibition of HCC, which is because free hGC33 and free SFB directly inhibit Wnt signal and Ras/Raf/MAPK, respectively. Such pathways can inhibit the proliferation of HCC cells to a certain extent. Although the nanoparticle-modified hGC33 (hGC33-null-NP) is connected to the nanosurface through chemical bonds, it did not affect hGC33′s targeting of GPC3 molecules and inhibition of Wnt activity. Nanoparticle-loaded SFB (SFB-NP), after being endocytosed by cells, was degraded to release SFB from the copolymer to inhibit the growth of HCC. In all, the inhibitory effect of hGC33-SFB-NP on HepG2 cell grafts was, as expected, more than on Huh-7 cell grafts, probably because HepG2 expresses GPC3 molecules.

The effect of hGC33-SFB-NP on xenotransplantation of HCC in nude mice and the changes of mice weight. Liver cancer cells were inoculated subcutaneously on the back of each nude mouse (n = 10). After 10 days, the tumor bearing mice were treated with PBS (control), free hGC33, free SFB, hGC33-null-NP, SFB-NP, and hGC33-SFB-NP. Tumor size (a , b ) and body weight (c , d ) of mice were monitored at designated time points
The body weight of nude mice in each treatment group also was measured, as shown in Fig. 9c, d. The body weight of the control group decreased gradually. The weight of mice in free hGC33, free SFB, SFB-NP, and hGC33-null-NP treatment groups also decreased progressively and not significantly less than in the control group. However, the weight of nude mice bearing HepG2 and Huh-7 treated with hGC33-SFB-NP only slightly decreased, and the weight remained relatively stable during the treatment cycle. These results support that the novel hGC33-SFB-NP nanodrug has no significant toxicity in nude mice, and the SFB loaded on the nanocarrier and the surface modified hGC33 can produce additive or even synergistic anti-tumor effect.
Discussion
To examine the suitability of hGC33-SFB-NP for targeted HCC therapy, we tested the model conjugates for their ability to bind to human glypican-3 on HCC cells in vitro; to inhibit glypican-3-positive HCC cell proliferation, migration, and Wnt/β-catenin signal transduction; and inhibit HCC that overexpress glypican-3 in vivo.
To covalently bind GPC3-specific antibody hGC33 with mal-PEG-b -PLGA nanoparticles, we cross-linked the free sulfhydryl group in the Fc segment of hGC33 with maleimide functionalized PEG-b -PLGA (mal-PEG-b -PLGA) by forming stable thioether bonds. Conjugation is a prerequisite for targeting of GPC3-positive HCC. A series of experiments, including the changes of nanoparticle diameter and zeta potential detected by lens and the intracellular uptake of hGC33-SFB-NP, verified the targeting of hGC33-SFB-NP to HepG2 (GPC3 + ) 细胞。 These results indicated that the binding activity of antibody hGC33 was not altered by the conjugation.
We directly detected the phagocytic effect of GPC3 + HepG2 and GPC3 − Li-7 cells on PEG-b -PLGA NP surface-modified hGC33 by confocal microscopy. After HepG2 and Li-7 cells were co-cultured with hGC33-coumarin 6-NP, the green signal intensity in HepG2 cells was significantly higher than in Li-7 cells, indicating that there were more nanoparticles in the HepG2 cells. This finding is consistent with the hGC33 antibody modified on the surface of PEG-b -PLGA NP specifically binding to glypican-3 on the surface of HCC cells and being internalized. The efficiency of hGC33-modified NP internalization depends on the expression level of GPC3 antigen on the cell surface.
We used the standard MTT assay to measure the efficiency of inhibiting the proliferation of hepatoma cells. Both hGC33-null-NP and hGC33 inhibited the growth of the GPC3-positive HCC cell line HepG2, but hGC33-null-NP and hGC33 did not affect the proliferation of GPC3-negative Li-7 cells (Fig. 3b). At the animal level, hGC33-null-NP or hGC33 alone inhibited the growth of Huh-7 and HepG2 xenografts to a certain extent, while hGC33-SFB-NP caused growth arrest of Huh-7 and HepG2 hepatoma xenografts in mice. The finding that hGC33-null-NP significantly inhibited GPC3-positive hepatoma cells showed that the inhibitory effect of PEG-b -PLGA NP surface-modified hGC33 on HCC cell proliferation depends on the expression of GPC3 antigen on the cell surface.
GPC3 regulates many pathways in HCC pathogenesis, including Wnt and YAP signaling [25,26,27]. GPC3 interacts with Wnt ligand and may be a coreceptor for Wnt and facilitate Wnt/Frizzled binding for HCC growth [28, 29]. We further examined the effect of nanodrug surface-modified hGC33 on Wnt signaling pathway in hepatoma cells. Like free hGC33, nanodrug surface-modified hGC33 inhibited the proliferation of hepatoma cells not only by blocking Wnt-induced signal transduction in HepG2 and Huh-7 cells of expressing GPC3, but also by inhibiting Wnt3a-induced β-catenin and YAP signal transduction. Previous studies have shown that YAP expression is regulated by β-catenin at the transcriptional level of HCC [30, 31]. In this study, free hGC33 and nanodrug surface-modified hGC33 inhibited Wnt3a-induced YAP activity, indicating that Yap/TAZ released from β-catenin complex can also initiate classic Wnt signaling transduction [32, 2]. These results indicate that typical Wnt and YAP cross talk through a variety of mechanisms. Compared with hGC33-null-NP and hGC33, hGC33-SFB-NP had stronger anti-proliferation and anti-migration ability in vitro and in vivo. Thus, hGC33 not only determines the specificity of HCC cells, but also increases the inhibitory effect of SFB on the proliferation and migration of HCC cells by blocking the key signals related to tumor growth, such as Wnt/β-catenin and Wnt/YAP signaling pathway.
Conclusion
Antibody hGC33 to glypican-3, a membrane protein that is overexpressed on hepatocellular carcinoma cells, increased binding of sorafenib-loaded polyethylene glycol-b-PLGA polymer nanoparticles (hGC33-SFB-NP) to glypican-3 on the cancer cells. Administration of the antibody-modified nanoparticles synergistically inhibited Wnt-induced signal transduction and Ras/Raf/MAPK signaling pathway; hepatocellular carcinoma cells were arrested in G0/1 phase by down-regulation of cyclin D1 expression, thus attenuating cancer cell migration by inhibiting epithelial–mesenchymal transition. hGC33-SFB-NP inhibited the growth of liver cancer in vivo and improved the survival rate of tumor-bearing mice.
数据和材料的可用性
Yes, all data have presented in the manuscript.
缩写
- AKT/PKB:
-
Protein kinase B
- c-MET:
-
HGFR:Hepatocyte growth factor receptor
- EE%:
-
Encapsulation efficiency %
- EMT:
-
Epithelial mesenchymal transition
- FTIR:
-
傅里叶变换红外光谱
- 1 H NMR:
-
1 H-nuclear magnetic resonance spectroscopy
- HCC:
-
Hepatocellular carcinoma
- LC%:
-
Drug loading %
- MAPK:
-
Mitogen-activated protein kinases
- PDI:
-
多分散指数
- PI3K:
-
Phosphoinositide 3-kinase
- pRAD51:
-
Phospho-RAD51
- SFB:
-
Sorafenib
- TEM:
-
透射电子显微镜
- YAP:
-
Yes-associated protein-1
纳米材料
- 用于癌症治疗的纳米粒子:当前的进展和挑战
- 钴掺杂 FeMn2O4 尖晶石纳米粒子的制备和磁性
- 纳米颗粒作为外排泵和生物膜抑制剂,恢复常规抗生素的杀菌效果
- Ag 纳米颗粒/BiV1-xMoxO4 与增强的光催化活性的协同效应
- 弹性刚度和表面附着力对纳米粒子弹跳的影响
- 银纳米结构的合成方法和应用的最新进展
- PEG 包覆的 CoFe2O4 纳米颗粒的毒性与姜黄素的处理效果
- 酸性胶溶剂对 TiO2 纳米颗粒锐钛矿-金红石比和光催化性能的影响
- 圆形金纳米粒子:粒径和浓度对拟南芥根系生长的影响
- 金属和金属氧化物纳米粒子的绿色合成及其对单细胞藻类莱茵衣藻的影响
- 纳米粒子和超声波控制的水过冷
- 氧化锌纳米粒子的特性及其对微生物的活性