

MoSe2-Ni3Se4 混合纳米电催化剂及其增强的析氢反应电催化活性
摘要
将 MoSe2 与其他过渡金属二硫属化物结合形成混合纳米结构是提高析氢反应 (HER) 电催化活性的有效途径。在这项研究中,通过种子诱导溶液法合成了具有花状形态的 MoSe2-Ni3Se4 杂化纳米电催化剂。 Ni3Se4 组分不是独立成核形成单独的纳米晶体,而是倾向于在 MoSe2 的超薄纳米薄片表面成核和生长,形成混合纳米结构。制备了具有不同 Mo:Ni 比例的 MoSe2-Ni3Se4 杂化纳米电催化剂,并比较了它们的 HER 催化活性。结果表明 HER 活性受 Mo:Ni 比例的影响。与纯 MoSe2 相比,Mo:Ni 摩尔比为 2:1 的 MoSe2-Ni3Se4 杂化纳米电催化剂表现出增强的 HER 性能,在 10 mA/cm 2 下的过电位为 203 mV 和每十倍频程 57 mV 的 Tafel 斜率。 MoSe2-Ni3Se4 杂化样品还观察到电导率提高和周转频率 (TOF) 增加。
介绍
传统化石燃料是我们社会的主要能源;然而,它们是不可再生和不可持续的,对环境造成严重污染。在替代能源中,氢能因其超高的能量密度而被认为是最有前途的清洁能源之一[1]。迄今为止,大规模生产氢气仍主要来自化石燃料来源[2]。煤气化和甲烷蒸汽重整工业生产 95% 的氢气 [3]。析氢反应 (HER) 被认为是一种有前途的生产高纯度氢气的途径 [1, 4, 5]。然而,酸性介质中 HER 的最佳电催化剂仍然是 Pt 基和其他贵金属材料 [6]。由于其稀缺性和高成本,Pt 基材料不适合用于大规模析氢 [7]。过渡金属二硫属化物 (TMD),如 MoS2、MoSe2、WS2 和 WSe2,受到了广泛关注。由于其优异的电化学性能和地球丰富的性质。作为典型的层状 TMD 半导体材料,MoSe2 具有与石墨相似的结构,由 Se-Mo-Se 层通过范德华力键合形成。此外,MoSe2 比 MoS2 更具金属性,并且在 MoSe2 边缘吸附氢的吉布斯自由能比 MoS2 低,从而导致更高的氢吸附[8]。因此,MoSe2及其杂化物作为HER电催化剂备受关注。
众所周知,只有活性位点对 HER 有效。对于像 TMD 纳米片这样的二维层状纳米结构,HER 的活性位点位于纳米片边缘 [9],而基底表面是惰性的。电催化剂的导电性也是 HER 的一个重要问题。作为一种半导体,与贵金属相比,MoSe2 较差的电子传输能力仍然限制了其在 HER 中的性能 [10]。因此,提高 TMD 催化剂活性的一般策略是提高电导率 [11, 12] 和增加活性位点数 [12,13,14]。同时,通过集成不同类型的半导体材料,尤其是具有优选取向的 TMD 来设计混合结构,被认为是调整半导体材料电子特性的重要方法 [15,16,17]。具有高效异质界面的混合纳米结构可以促进快速的界面电荷转移,这对电化学反应至关重要 [18]。此外,众所周知,在电化学反应过程中产生氢气需要三个基本步骤,即吸附、还原和解吸[19]。由不同化学成分组成的杂化材料的优势之一是它们可以突破许多单组分催化剂对所有三个中间反应过程都无效的限制。最近,一些研究人员通过使用不同的方法将 Ni 基催化剂与各种形态的 MoSe2 结合,以提高 HER 性能 [15, 18, 20]。 MoSe2 与 Ni 硒化物结合形成杂化结构可以利用两种异质组分之间相互作用产生的协同效应来提高电催化活性。例如,DFT 计算表明 MoS2(1-x )Se2x /NiSe2 在 (100) 和 (110) 平面上的氢吸附吉布斯自由能比纯 MoS2(1−x )Se2x ,这可能导致活性位点的氢覆盖率更高,从而实现出色的电催化性能[21]。
在此,我们尝试通过在花状 MoSe2 种子的表面生长 Ni3Se4 来制备混合纳米电催化剂,这些种子是通过我们之前研究中报道的胶体法合成的 [22]。这种种子诱导生长方法为构建各种 TMD 混合纳米结构提供了一种简便的方法。我们选择 Ni3Se4 作为混合组件的原因是 Ni3Se4 比其他硒化镍具有更高的导电性 [23]。为了研究 Ni3Se4 对催化剂活性的影响并找出最佳组成比,我们系统地调节 Ni3Se4 和 MoSe2 的含量,发现在 MoSe2-Ni3Se4 混合体系中加入适量的 Ni3Se4 可以提高 HER 性能.研究结果表明,构建MoSe2-Ni3Se4混合纳米结构是提高纯MoSe2的HER性能的有效途径。
方法/实验
MoSe2–Ni3Se4 杂化纳米电催化剂的合成
MoSe2-Ni3Se4 杂化纳米电催化剂的合成包括两个步骤。在第一步中,根据我们之前研究中报道的方法合成 MoSe2 种子 [22]。简而言之,将 10 mL 油酸(OA,85%,Aladdin Bio-Chem Technology Co., Ltd.)和 0.4 mmol 六羰基钼(Mo(CO)6, 98%,J&K Scientific Ltd.)混合并加热在氩气中缓慢升温至 85 °C。随后,将混合溶液的温度升至 200 °C,加入 6.7 mL 含有 1-十八碳烯(ODE,90%,Aladdin Bio-Chem Technology Co., Ltd.)和 Se(99.999%,J&K Scientific Ltd.) 以 0.15 mmol/mL 的 Se 浓度以 0.5 mL/min 的注射速度注射到反应溶液中。注入完成后,反应继续保持 30 分钟以生成 MoSe2 种子。在下一步中,将反应温度升至 300 °C,将 3.3 mL ODE 和 Se 的混合物与乙酰丙酮镍(II)(Ni(acac)2, 0.2 mmol, 96%, J&K Scientific Ltd.)混合。 ) 注入反应混合物并在 300 °C 下保持 30 分钟。冷却至室温后,反应产物用乙醇和己烷洗涤,然后在室温下干燥。合成的样品标记为 Mo2Ni1,表示 MoSe2-Ni3Se4 混合样品中 Mo:Ni 的摩尔比为 2:1。除了在反应中加入不同质量的Ni和Se源混合物外,其他MoSe2-Ni3Se4纳米杂化物样品采用相同的方法合成。
特征化
使用X射线衍射仪(Bruker D8-Advance)表征晶相。使用 JEM-2100 透射电子显微镜获得透射电子显微镜 (TEM) 图像。使用 TECNAI F-30 透射电子显微镜进行高角度环形暗场 (HAADF) 成像和相应的元素映射。使用 SU-70 扫描电子显微镜获取扫描电子显微镜 (SEM) 图像。 X射线光电子能谱(XPS)数据通过Al Kα源的光谱仪(PHI QUANTUM 2000)获得。
电化学测试
电化学测试在包含 Ag/AgCl 参比电极、石墨棒对电极和玻璃碳工作电极的标准测试系统中进行,这些电极连接到使用 H2SO4 (0.5 M) 作为电解质的 Autolab 302N 电化学工作站。为了制备电催化剂墨水,将合成的电催化剂(4 mg)、科琴黑炭黑(0.5 mg)和 Nafion 溶液(30 μL)与乙醇含量为 20 vol% 的乙醇-水溶液(1 mL)混合。然后将混合物超声处理30分钟。最后,将 5 μL 墨水(含约 20 μg 电催化剂)沉积在玻碳电极上,形成负载量约为 0.286 mg/cm 2 的薄膜 并在室温下干燥。极化曲线是通过使用2 mV s -1 的扫描速率获得的 在 25 °C 下从 0.2 到 − 0.6 V(相对于可逆氢电极 (RHE))。电化学阻抗谱 (EIS) 数据是在 0.01 Hz 到 100 kHz 的频率范围内在 – 260 mV 下获得的。通过循环伏安法(CV)测试获得0.1~0.2 mV的双电层电容(非法拉第电位)并计算电极的有效表面积。
结果与讨论
MoSe2-Ni3Se4 杂化纳米电催化剂的合成基于种子诱导策略,其中纳米级 Ni3Se4 在预先形成的 MoSe2 种子上原位生长(图 1)。在第一步中,在 ODE 中,在 200 °C 下,在 OA 存在下,通过 Mo 前体(Mo(CO)6)和 Se 之间的反应合成 MoSe2 晶种,其中进一步在加热过程中形成超薄的 MoSe2 纳米薄片自组装成花状 MoSe2 颗粒 [22]。具有大表面积的花状形态可以促进第二组分的分散和密切相互作用[24]。在温度达到 300 °C 后,将含有 Ni(acac)2 和 ODE-Se 的溶液快速注入含有 MoSe2 晶种的热反应混合物中。在这个阶段,Ni3Se4 在 MoSe2 纳米薄片表面成核和生长,形成 MoSe2-Ni3Se4 混合纳米结构。这种简便的合成策略可有效地在相似的实验条件下合成具有不同Mo:Ni比的MoSe2-Ni3Se4杂化纳米电催化剂,并可用于构建其他MoSe2基杂化纳米电催化剂。
<图片>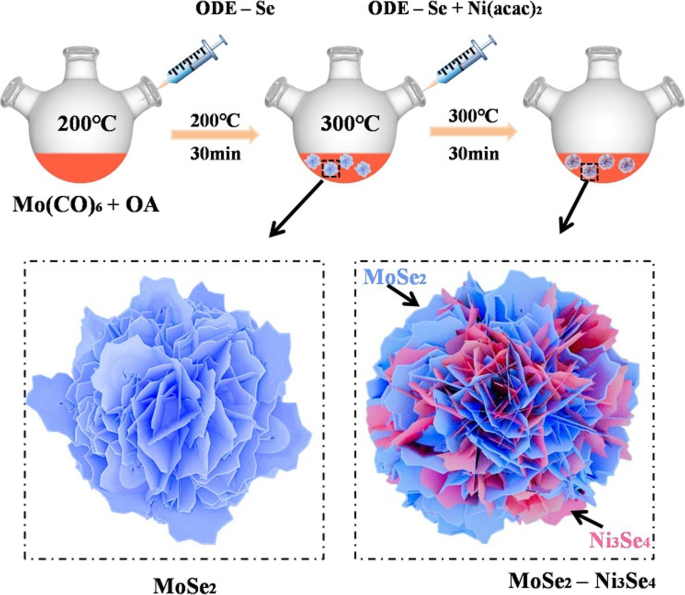
MoSe2-Ni3Se4杂化纳米电催化剂形成示意图
图 2 比较了纯 MoSe2 和 MoSe2–Ni3Se4 混合样品的 XRD 谱。纯 MoSe2 样品的衍射峰与六方 MoSe2 (PDF#29-0914) 一致,而不同 Mo:Ni 比的 MoSe2-Ni3Se4 杂化样品表现出六方 MoSe2 和单斜 Ni3Se4 的组合峰 (PDF#13-0300) .随着Ni前驱体含量的增加,XRD图中Ni3Se4的峰强度也增加,这表明MoSe2-Ni3Se4杂化纳米电催化剂中Ni3Se4的浓度也增加。因此,可以通过控制添加的 Ni 前驱体的含量来调节 MoSe2-Ni3Se4 混合纳米电催化剂中 Ni3Se4 的含量。 SAED 分析(附加文件 1:图 S1)还揭示了六方 MoSe2 和单斜 Ni3Se4 的共存,这证实了 XRD 结果。随着Ni前驱体含量的增加,属于Ni3Se4的衍射环也变得突出,表明MoSe2-Ni3Se4杂化纳米电催化剂中Ni3Se4组分的相对含量也增加。
<图片>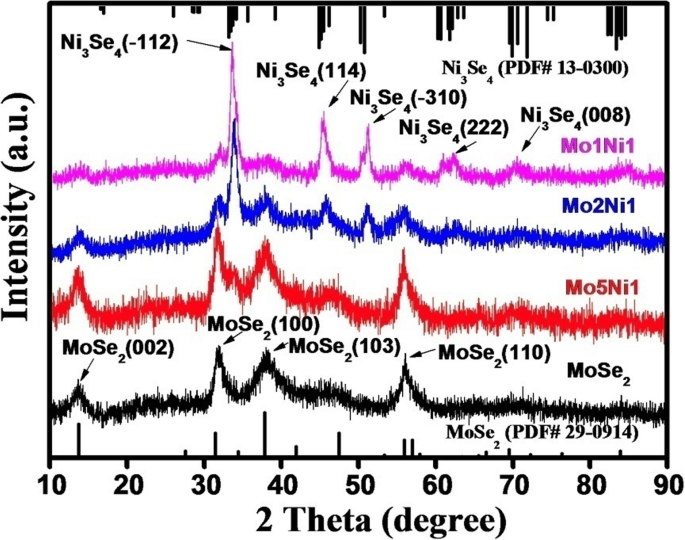
具有不同 Mo:Ni 比例的纯 MoSe2 和 MoSe2–Ni3Se4 混合样品的 XRD 谱。还包括块体 MoSe2 和 Ni3Se4 的参考图案
通过 SEM 和 TEM 分析所制备样品的形貌。纯 MoSe2 具有花状形态,尺寸范围为 100 至 200 nm(附加文件 1:图 S2)。在加入 Ni3Se4 后,可以清楚地看到纳米花的花瓣开始变厚(图 3),随着 Ni3Se4 含量的增加,花状形态趋于逐渐消失。 Mo2Ni1 样品的高分辨率 TEM(HRTEM)分析(图 4a、b)揭示了两种明显的晶格条纹:一种具有 0.64 nm 的面间距对应于 MoSe2 [25] 的 (002) 平面,而面间距为 0.27 nm 的一个与 Ni3Se4 的 (-112) 面非常吻合。结果证实了混合纳米结构中同时存在 MoSe2 和 Ni3Se4 组分,并且纳米花瓣的主表面由 MoSe2 的 {001} 面构成。此外,两种不同的晶格条纹大致平行,表明Ni3Se4可能沿MoSe2的c轴生长在MoSe2的{001}面上。
<图片>
SEM 图像 (a , b , d , e , g , 和 h ) 和 TEM 图像 (c , f , 和 i ) Mo5Ni1 (a -c ), Mo2Ni1 (d -f ) 和 Mo1Ni1 (g -i ) 样品
<图片>
HRTEM 图像 (a 和 b ), HAADF 图像 (c ) 和元素图 (d -f ) Mo2Ni1 样品
能量色散 X 射线光谱 (EDS) 元素图以及 HAADF 图像(图 4d -f ) 证实了 Se、Ni 和 Mo 的存在。然而,Mo 和 Ni 的空间分布略有不同。 Mo 基本上均匀分布在纳米花中,而 Ni 往往集中在纳米花的花瓣附近,这表明 Ni3Se4 应该在 MoSe2 花瓣上生长。在 MoSe2 上覆盖较厚的 Ni3Se4 层可能会阻塞 MoSe2 的活性位点,最终导致 HER 性能下降。除了 Ni 和 Se 源的注入量,注入速率也影响 MoSe2-Ni3Se4 杂化纳米结构的形貌。当使用较小注入速率 (1.65 mL/min) 的 Ni 和 Se 源时,结果表明产物具有不均匀的形态(附加文件 1:图 S3)。这表明MoSe2-Ni3Se4杂化纳米结构的形成也是一个动力学控制的过程。
XPS 分析(图 5a-d)进一步验证了混合样品中 Mo、Ni 和 Se 的存在(以 Mo2Ni1 为例)。对于 Se 3d 区域(图 5b),54.75 eV 和 55.75 eV 处的两个峰分别归属于 Se 3d5/2 和 Se 3d3/2,这表明 Se at 的氧化态为 - 2 [26]。 59.37 eV 处的明显峰值表明表面的 Se 物质已被氧化 [20, 26]。在图 5c 中,位于 229.37 和 232.50 eV 的两个峰分别归属于 Mo 3d5/2 和 3d3/2,表明 Mo 的 +4 氧化态 [8, 11, 26]。在图 5d 中,Ni 2p 峰清晰可见,856.62 和 874.12 eV 处的峰分别与 Ni 2p3/2 和 Ni 2p1/2 吻合。 861.87 和 880.37 eV 的两个卫星峰表明 Ni 处于接近 +2 的氧化态 [27]。
<图片>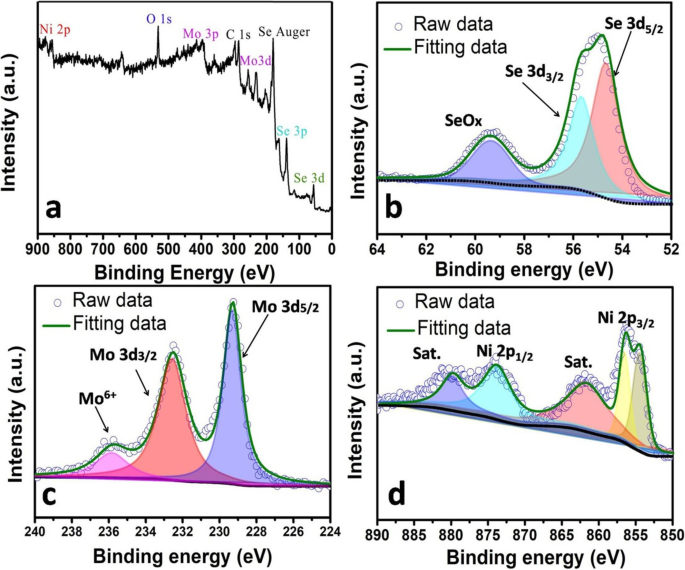
Mo2Ni1 样品的 XPS 光谱。 一 是调查频谱。 b ), c , 和 d 分别显示Se、Mo和Ni的扩展光谱
从上述表征结果可以理解 MoSe2-Ni3Se4 杂化纳米结构的形成机制。花状的 MoSe2 种子在诱导 MoSe2 表面形成 Ni3Se4 中起重要作用。在300℃的反应温度下,Ni(acac)2容易分解与Se反应生成Ni3Se4。 MoSe2 的表面可以作为异质成核位点来诱导 Ni3Se4 的成核。显然,这种异相成核过程比均相成核需要更少的活性能量。因此观察到 Ni3Se4 在 MoSe2 表面生长形成花瓣状形态,而不是通过独立均匀成核形成的分离颗粒。随着Ni和Se源量的进一步增加,Ni3Se4倾向于在已经形成的Ni3Se4花瓣表面生长。结果,观察到了 Ni3Se4 花瓣厚度增加的 MoSe2-Ni3Se4 杂化纳米结构(见图 3 所示的形态演变)。
在酸溶液中使用三电极系统测量所制备催化剂的电催化活性。如图 6a 所示,所有起始过电位(即达到 1 mA cm -2 电流密度所需的电位) ) [28] 各种催化剂都很少。 Mo5Ni1 样品要求 HER 的最低起始过电位为 128 mV,而对于其他催化剂,MoSe2、Mo2Ni1、Mo1Ni1 和 Ni3Se4 的起始过电位值分别为 163、140、162 和 216 mV。当阴极电流密度达到-10 mA cm -2 ,Mo2Ni1 样品需要 203 mV 的最小过电位。 MoSe2、Mo5Ni1、Mo1Ni1 和 Ni3Se4 所需的过电位分别为 234、220、250 和 299 mV。为了进一步研究获得的样品,使用 Tafel 方程分析了 Tafel 曲线的线性部分:
$$ \eta =b\;\log\;j+a $$ (1)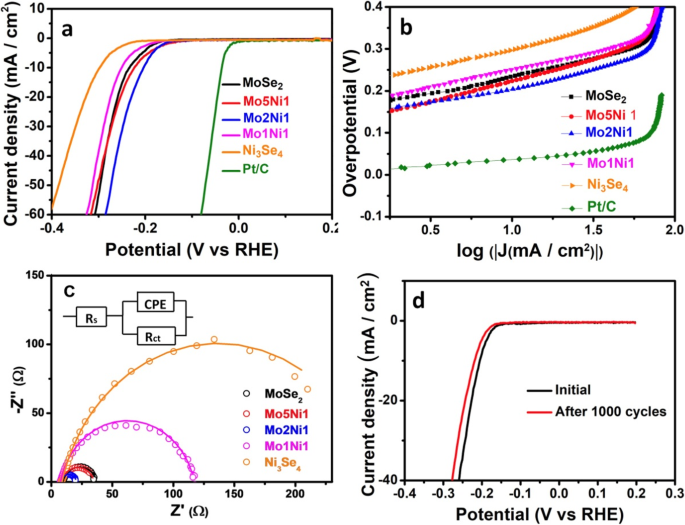
极化曲线 (a ) 和相应的 Tafel 图 (b ) MoSe2、Mo5Ni1、Mo2Ni1、Mo1Ni1、Ni3Se4 和 Pt/C。 c 奈奎斯特在 250 mV 的过电位下绘图。 d Mo2Ni1样品1000次循环前后的极化曲线
其中 j 是电流密度,η 是过电位,b 是塔菲尔斜率。从图 6b 中可以看出,Mo2Ni1 样品的 Tafel 斜率为 57 mV/十倍频程。该值远小于 Mo5Ni1(85 mV/十年)、Mo1Ni1(88 mV/十年)、Ni3Se4(82 mV/十年)和 MoSe2(71 mV/十年)样品的斜率。同时,Pt/C 的 Tafel 斜率约为 33 mV/十年,与已知值相当[29]。理论上,较低的 Tafel 斜率表明较快的 HER 动力学 [30]。 Tafel 斜率可以揭示 HER 过程中的主要反应机制 [15, 19]。有三个主要步骤可以参与HER过程,即Volmer反应:H + (aq) + e - → Hads, Heyrovsky 反应:Hads + H + (aq) + e - → H2 (g),和 Tafel 反应 Hads + Hads →H2 (g)。在 25 °C 下,三个反应的 Tafel 斜率值分别为 118 mV/十年、39 mV/十年和 29 mV/十年 [19]。因此,我们的研究结果表明,Volmer-Heyrovsky [31,32,33] 的机制应该在 HER 中所有制备的样品中占主导地位。
为了进一步研究电极的动力学,EIS 获得的五个样品的奈奎斯特图如图 6c 所示。电荷转移电阻 (R ct)是从低频区域实现的,与电极的动力学密切相关。 R 的较小值 ct 与更高的反应速率有关 [34]。 R 的价值 Mo2Ni1 的 ct 为 13.0 Ω,这是五个样品中的最低值。对于其他样本,R MoSe2、Mo5Ni1、Mo1Ni1 和 Ni3Se4 的 ct 值分别为 27.5、27.1、109.1 和 254.6 Ω。最低的 R Mo2Ni1 的 ct 表明所制备的样品中电荷转移过程最快。结果进一步证明了 Mo2Ni1 样品优异的 HER 电催化效率。更好的导电性可能是由于 MoSe2 和 Ni3Se4 之间的协同效应对电子结构的调制。图 6d 给出了表征 Mo2Ni1 样品稳定性的极化曲线。 1000次循环后,催化性能仅表现出轻微下降。协同效应在控制催化表面上的吸附-吸收相互作用方面起着重要作用,从而决定了催化反应的速率决定步骤 [35]。因此,利用协同效应构成了杂化纳米结构增强HER活性的主要优势。
为了粗略计算催化剂的电化学活性表面积 (ESCA),电化学双层电容 (C dl) 是使用循环伏安法 (CV) 在不同扫描速率下测量的(附加文件 1:图 S4)。 Δj 的图 =(j a-j c)( j a 和 j c 分别是在 0.15 V 电压下充电和放电时的电流密度)与扫描速率的关系如图 7a 所示,C dl 值被计算为斜率的一半。 Mo2Ni1 表现出 C dl 值为 2.67 mF cm −2 略小于该值 (3.06 mF cm −2 ) MoSe2 和 Mo5Ni1 (2.82 mF cm −2 ),表明 Ni3Se4 的加入不能进一步增加电化学活性表面积,其结果与 TEM 观察结果一致。因此,Mo2Ni1 样品的 HER 催化活性提高的原因不太可能是由于电化学活性表面积的增加,而是 MoSe2 和 Ni3Se4 之间的协同作用,以及电导率的提高。此外,我们估计了各种催化剂的活性位点数量和周转频率(TOF)。活性物质的数量由不同催化剂的 CV 曲线获得,该曲线在磷酸盐缓冲盐水电解质中从 - 0.4 到 0.6 V 记录,扫描速率为 50 mV s -1 (附加文件 1:图 S5)[30, 36]。 Mo2Ni1 的计算活性位点数为 1.02 × 10 −6 mol 而 MoSe2 的为 0.77 × 10 −6 摩尔。此外,Mo2Ni1 的每个活性位点在 – 200 mV 处的计算 TOF 为 3.4 s -1 , 也大于 (2.1 s −1 )的 MoSe2(图 7b)。从理论上讲,催化剂的 HER 活性可归因于三个因素:(a)活性位点数量,(b)活性位点质量(周转频率)和(c)活性位点之间的电导率 [37]。在这项工作中,虽然与 MoSe2 相比,Mo2Ni1 的 Cdl 值略小,但它具有最低的电荷转移阻抗、最多的活性位点和最高的 TOF。因此,它表现出最好的整体HER活性。
<图片>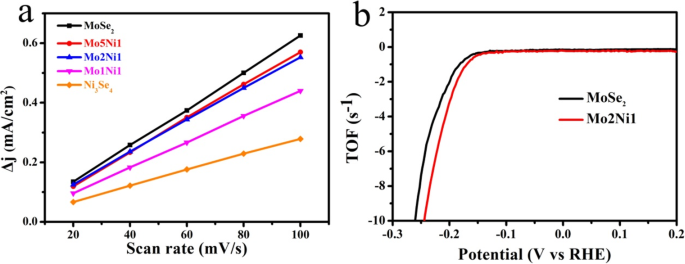
一 MoSe2、Mo5Ni1、Mo2Ni1、Mo1Ni1 和 Ni3Se4 样品的双层容量电流与扫描速率的关系。 b 纯 MoSe2 和 Mo2Ni1 样品的计算 TOFs
结论
已经开发了一种用于合成 MoSe2-Ni3Se4 杂化纳米电催化剂的种子诱导溶液路线。具有由超薄纳米薄片组装而成的花状形态的 MoSe2 种子已被用于诱导 Ni3Se4 在 MoSe2 花瓣上的生长。 MoSe2-Ni3Se4 杂化纳米电催化剂的化学成分可以通过调节 Ni3Se4 的含量来调节。据观察,Ni3Se4 与 MoSe2 结合形成混合纳米结构可以提高 MoSe2 的 HER 性能。 Mo:Ni 比为 2:1 的 MoSe2–Ni3Se4 杂化纳米电催化剂提供了卓越的 HER 性能,其起始过电位为 140 mV,在 10 mA cm −2 下的过电位为 201 mV 和 57 mV dec −1 的小塔菲尔斜率 在酸性条件下。还观察到了改进的电导率和TOF。
数据和材料的可用性
不适用。
缩写
- 她:
-
析氢反应
- XRD:
-
X射线衍射
- TMD:
-
过渡金属二硫属化物
- SEM:
-
扫描电镜
- TEM:
-
透射电子显微镜
- SAED:
-
选区电子衍射
- HAADF:
-
高角度环形暗场
- XPS:
-
X射线光电子能谱
- EIS:
-
电化学阻抗谱
- TOF:
-
周转频率
- R ct:
-
电荷转移电阻
- C dl :
-
电化学双层电容
纳米材料
- 用于增强药物递送的纳米纤维和细丝
- 用于改进诊断和治疗应用的多功能金纳米粒子:综述
- 具有可控厚度的二硫化钼用于电催化析氢
- Sb/坡缕石 (PAL) 纳米颗粒的制备和增强催化氢化活性
- 水热合成 In2O3 纳米颗粒混合孪晶六边形圆盘 ZnO 异质结构以提高光催化活性和稳定性
- 用于高效光催化制氢的 ZnO@TiO2 空心球的分级异质结构
- PPy 包覆的 MnO2 混合微材料的制备及其作为锂离子电池阳极的改进循环性能
- Cu2ZnSnSe4 纳米片的一锅法合成及其可见光驱动的光催化活性
- Fe3+ 的可恢复荧光探针 BHN-Fe3O4@SiO2 混合纳米结构及其在生物成像中的应用
- PtNi 合金助催化剂改性曙红 Y 敏化 g-C3N4/GO 杂化物以实现有效的可见光光催化产氢
- 通过将 Cd0.5Zn0.5S QD 加载到 Ni2P 多孔纳米片上来增强光催化产氢
- 氧化锌纳米粒子的特性及其对微生物的活性