

钒原子在清洁和石墨烯覆盖的 Cu(111) 表面吸附的电子特性
摘要
已经使用从头算理论方法系统地研究了吸附在清洁和石墨烯覆盖的 Cu(111) 表面上的钒原子的电子性质。在这项工作中考虑了钒吸附的两种覆盖范围(1/9 ML 和 1 ML)。我们的计算表明,在 V/Cu(111) 的上述两个覆盖范围内,发现留在 Cu 表面下方的 V 是最稳定的吸附位点。然而,这种吸附可能导致不希望的特性。因此,我们引入石墨烯作为缓冲层,以有效减轻 V 和 Cu 表面之间的直接相互作用。计算表明,原始石墨烯层的电子特性受 C 原子与 V 吸附原子相互作用的显着影响;结果,石墨烯的狄拉克点在两个覆盖范围内都被“破坏”了。在 V/Gra/Cu(111) 系统中,石墨烯层与基底 Cu 原子之间的相互作用与 Gra/Cu(111) 系统一样仍然很弱。此外,1/9 ML 的相对较低的覆盖范围会产生自旋极化系统,而在 1 ML 的覆盖范围内观察到非自旋极化系统。这一发现为钒基材料在现实中的应用提供了一条新途径。
背景
多相催化在化学和能源工业的许多领域都起着至关重要的作用。目前,大量研究集中在理解、改进和设计新催化剂上。过渡金属原子在贵金属底物上的吸附会影响相应的催化性能,这是催化领域最重要的课题之一 [1,2,3,4,5,6,7]。特别是,一种单层金属在金属表面的吸附在各种吸附系统中表现出显着不同的化学和催化性能 [5,6,7]。一般来说,材料的催化性能取决于它们的原子结构、组成和费米能级附近的电子态 [8,9,10,11,12]。预计基材会直接和/或间接影响金属沉积物的催化性能。众所周知,Cu(111)表面是过去几十年来研究最彻底的单晶金属表面之一 [13,14,15,16,17,18,19,20,21,22,23,24 ]。特别是在过去的十年中,Cu(111) 表面被认为是通过化学气相沉积 (CVD) 生长高质量和大面积石墨烯的主要基材 [22,23,24]。石墨烯的新电子特性可以很好地保留在这样的基板上。 Cu(111)表面后期4d过渡金属(如Rh[25]、Pd[26,27,28,29,30]、Ir[31]和Pt[29,32,33])的吸附在实验和理论上都得到了广泛的研究。然而,对吸附在Cu(111)表面的早期3d过渡金属原子的研究相对缺乏[34,35,36,37]。在这里,我们关注早期的 3d 过渡金属元素钒,因为它具有生化相关性和在多个工业领域的广泛应用,例如多相催化、分子网络、纳米材料和电池制造 [38]。由于其价态灵活,钒基聚阴离子材料被提议作为替代商业正极材料 LiCoO2 和 LiMn2O4 的候选材料 [39]。因此,研究钒原子的吸附特性可以促进其在现实中的应用。可以预期所研究系统的可能应用如下。 (1)钒的常见氧化态可以是+ 2、+ 3、+ 4和+ 5;因此,它可以在纳米材料工业中用作强大而多功能的催化剂[38]。 (2) 金属态的钒可用于催化 CO 歧化为 C 和 CO2 [40]。 (3) 分析吸附在自由电子浓度较弱的表面上的 TM(即钒)原子也很有趣,因为可能会增加电和热传导 [41]。此外,人们对可用于记录介质、磁性墨水和自旋电子器件的二维表面系统中的磁序引起了极大的兴趣。
在这项工作中,我们报告了基于密度泛函理论 (DFT) 的钒原子在清洁 Cu(111) 表面和石墨烯覆盖的 Cu(111) 表面上的吸附的系统研究。对于上述两个系统,考虑了钒吸附原子的两个对比覆盖率(即 1/9 ML 和 1 ML)来评估覆盖率对电子和磁性的影响。无论 V 覆盖率如何,吸附在清洁 Cu(111) 表面上的 V 的最低能量吸附位点都在表面下方而不是表面上方。对于 V 在石墨烯覆盖的 Cu(111) 表面上的吸附,吸附位点依赖于覆盖率,即具有最大配位的空心位点在 1/9 ML 覆盖率上有优势,而具有低配位的顶部位点首选 1 ML 覆盖率。同时,对于 V/Cu(111) 和 V/Gra/Cu(111) 系统,V 吸附原子的自旋极化在 1/9 ML 覆盖范围内在能量上受到青睐,而在 1 ML 覆盖范围内没有发现磁性。此外,对于 1/9 ML V/Gra/Cu(111) 系统,石墨烯 C 原子的净磁矩约为 0.16 μB/每个碳,这与 Gra/Cu(111) 系统中的结果不同。为了深入了解 V/Cu(111) 和 V/石墨烯/Cu(111) 系统中的相互作用,详细分析了费米表面的电子态。总之,我们的研究有助于理解V/Cu(111)和V/Gra/Cu(111)体系的电子特性。
方法
我们的计算是通过采用 Vienna ab initio 模拟包 (VASP) [42] 进行的,该包基于自旋极化密度泛函理论 [43]、平面波基础和投影增强波 (PAW) 表示 [44] ]。计算中采用了广义梯度近似 (GGA) 中的 Perdew-Burke-Ernzerhof (PBE) 交换相关能量泛函 [45](一些使用 B3LYP [46, 47] 和 HSE06 [48] 混合泛函的比较研究也是必要时提出)。为了准确描述石墨烯和Cu(111)表面之间的范德华(vdWs)相互作用,采用具有vdWs校正(DFT-D2)[49]的PBE功能。截止平面波动能设置为 500 eV。 Cu(111) 表面通过使用包含七个 Cu 层以及大约 20 Å 的真空间距的板条模型进行建模。 Cu(111) 和 Gra/Cu(111) 表面上不同的 V 覆盖率通过使用不同的超胞进行建模。对于 1/9 ML 和 1 ML 的 V 覆盖率,我们分别采用了 (3 × 3) 和 (1 × 1) 表面晶胞。 Monkhorst-Pack 方案 [50] 具有 24 × 24 × 1 k- 网格用于采样 (1 × 1) 表面晶胞的布里渊区积分,而 8 × 8 × 1 k- 网格用于(3 × 3)表面晶胞。在优化过程中,板坯最下面的三个铜层被冻结,而系统的其余原子完全松弛,直到每个原子上的力小于 0.01 eV/Å。钒原子被吸附在板坯的一侧。由于基于我们的计算发现的能量校正可以忽略不计,因此本研究未考虑偶极校正[51]。
结果与讨论
钒原子吸附在清洁的 Cu(111) 表面
在本节中,我们在两个覆盖范围(即 1/9 ML 和 1 ML)下直接在干净的 Cu(111) 表面上呈现 V 吸附的结果。为了在 Cu(111) 表面找到 V 原子的有利吸附位点,每个覆盖范围都考虑了七个可能的吸附位点,即 top、fcc、hcp、subT、top-fcc、top-hcp 和桥位,如图1a所示。特别需要注意的是,subT是Cu表面下方的位点,V吸附原子与表层Cu原子交换位置(并将Cu原子移动到钒原子正上方的位点);参见图 2a、b。对于两种吸附覆盖,所有七个吸附位点的吸附能都是为 V/Cu(111) 系统计算的。得到的结果如图 1b、c 所示。这里,每个钒原子的吸附能(Ead)由下式计算:
$$ {\mathrm{E}}_{\mathrm{ad}}=\left[\left({NE}_V+{E}_{Cu(111)}\right)-{E}_{V/ Cu (111)}\right]/N $$其中 E V 是孤立钒原子的能量,E 铜 (111) 是所涉及的清洁 Cu(111) 表面的总能量,E V /铜 (111) 是 V/Cu(111) 系统的总能量,N 是涉及的 V 原子数。从图 1b、c 中可以看出,对于上述覆盖范围,在 Cu(111) 表面上吸附 V 时,subT 位点在能量上是有利的。在此基础上,我们将在以下讨论中仅考虑 subT 站点。计算的吸附能、V原子与其相邻Cu原子之间的键长以及V/Cu(111)吸附原子的原子磁矩列于表1中。从表1中可以看出,吸附能对于 1/9 ML 和 1 ML 的覆盖范围,每个 V 原子的 Ead 分别为 2.17 和 1.61 eV,这表明 V 原子与 Cu(111) 表面的相互作用非常强。此外,吸附能随着 V 覆盖率的增加而降低,这意味着 V-V 相互作用变强,而 V 层与 Cu 表面之间的相互作用变弱。对于 1/9 ML 和 1 ML 的覆盖范围,V 原子与其相邻的 Cu 原子之间的最短键长分别为 2.27 和 2.37 Å。这意味着对于 1/9 ML,V 吸附原子和 Cu 底物之间的相互作用相对较强,这与吸附能的计算结果一致。计算中还考虑了V吸附原子的铁磁(FM)阶次,FM阶次的自旋极化能量由ΔE计算 =(E 没有 _mag − E 调频 )/N (与 E 没有 _mag 是非磁性状态的能量)。对于 1/9 ML 覆盖范围(见表 1),钒原子的自旋极化能量为 110 meV,而对于 1 ML 覆盖范围则没有磁性。对于钒的 1/9 ML 覆盖范围,V 的原子磁矩为 1.34 μB,这与气相 V 原子的值 (3 μB) 有很大不同。关于这一点,我们稍后再讨论。
<图片>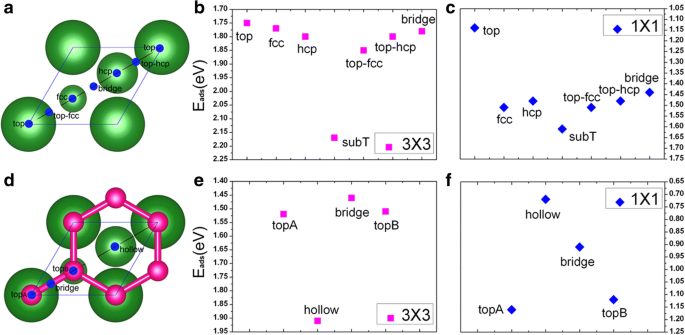
一 干净的Cu(111)(1 × 1)表面的吸附位点:最大的球表示表面Cu原子,较小的球表示亚层Cu原子; b , c 分别为 1/9 ML 和 1 ML 覆盖度下 V 原子在 Cu(111) 上的不同吸附位点的吸附能; d 石墨烯覆盖的Cu(111)(1 × 1)表面,红球表示石墨烯的C原子; e, f V原子在石墨烯覆盖的Cu(111)上的吸附能分别为1/9 ML和1 ML
<图片>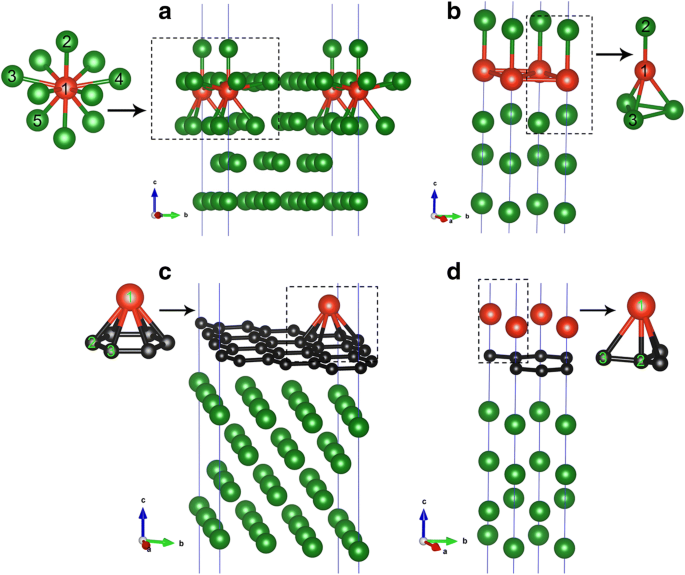
a 的 V/Cu(111) 系统的几何形状 1/9 ML 和 b 1 ML 覆盖。 c 的 V/Gra/Cu(111) 系统的几何形状 1/9 ML 和 d 1 ML 覆盖 V 原子。红、黑、绿球分别代表V、C、Cu原子
接下来,我们讨论 V/Cu(111) 系统的电子结构。 V 在 Cu(111) (3 × 3) 和 Cu(111) (1 × 1) 表面(即 1/9 和 1 ML)上吸附的能带结构如图 3 所示,其能带结构如图 3 所示。还绘制了相应的清洁 Cu(111) (3 × 3) 和 Cu(111) (1 × 1) 表面以进行比较。图 3a、d 均可用于讨论清洁 Cu(111) 的电子结构;我们在这里选择图 3d。我们发现 s 电子和 d Cu 的电子有助于清洁 Cu(111) 表面的系统电导。更详细地,我们在图 3d 中标记了费米面上的代表点(A、B、C、D、E)。 A点和E点主要来自d yz 表面铜原子的电子。 B 点和 C 点显示了 d 的贡献 xy 和 d x 2 − y 2 分别为 Cu 原子的电子。 D点描述了s的混合 带有 d 的电子 z 2 和 d x 2 − y 2 相邻铜原子之间的电子。当 V 吸附在 Cu(111) 上时,随着 V 覆盖率的变化,获得的能带结构以不同的方式变化。对于 1/9 ML 覆盖范围(如图 3b、c 所示),自旋向上和自旋向下通道中的能带结构不同,表明存在自旋极化特征。在图 3 中,红点表示 V 吸附原子的贡献,而银灰色点表示背景 Cu 的贡献。从自旋向上通道(即多数自旋),d V 吸附原子的电子和衬底 Cu 原子对费米表面的电子态有显着贡献。 d 的杂交 表面Cu原子和V吸附原子的电子清晰可见。为了清楚地描述,我们还在图 3b 中的费米面上标记了一些代表点(A、B、C、D)。其中,A点表示d的混合 z 2 具有 d 的 V 吸附原子的电子 yz , d z 2 表面铜原子的电子。所有 B、C 和 D 点均表示来自 d 的贡献 V 吸附原子的电子。例如,B 点仅展示了 d x 2 -y 2 V 吸附原子的电子。对于自旋向下通道(即少数自旋),能带结构表明由 V 吸附原子贡献的电子态都远高于费米能级(未占据)。对费米面的贡献主要来自s , d Cu 原子的电子,V 吸附原子的电子贡献很小。这两个自旋通道之间存在的差异表明 V 吸附原子上的磁矩 (1.34 μB)。对于 1 ML 覆盖(如图 3e、f 所示),与 1/9 ML 的情况不同,吸附系统是非自旋极化的。为方便起见,我们还在图 3e 中标记了费米面上的代表点(A、B、C、D、E、F、G、H)。 A点和H点都对应于d的贡献 yz Cu原子底层的电子。 B、D、E、F 和 G 点的电子态由 d 贡献 V 吸附原子的电子。例如,只有 d x 2 − y 2 和 d xz 的 V 吸附原子分别对 B 和 D 处的电子状态有贡献。对于费米面上 B 和 D 之间的那些点,我们发现了 d 的复杂混合 具有 s, d 的 V 吸附原子的电子 V 原子周围的 Cu 原子的电子。例如,对于C点,电子结构的特征在于s, d的混合 yz , 和 d z 2 dz 2 表层 Cu 和最顶层 Cu 原子的电子 V吸附原子的电子。
<图片>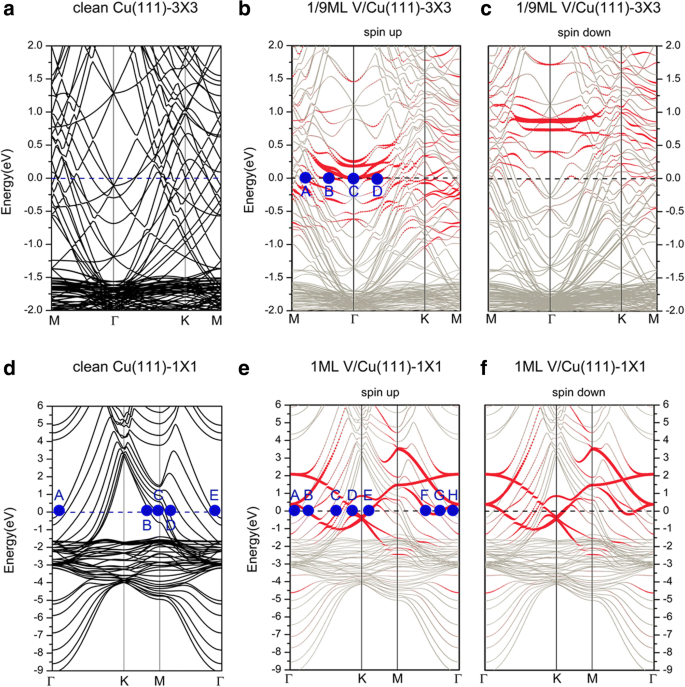
干净的 Cu(111) 表面的能带结构,绘制在 a 的布里渊区 (BZ) 中 3 × 3个单元格和d 1 × 1个单元格。 b覆盖1/9 ML时Cu(111)表面V吸附的能带结构 旋转并c 向下旋转。 e覆盖1 ML时Cu(111)表面V吸附的能带结构 旋转并f 向下旋转
对于 1/9 ML 和 1 ML 覆盖,V 在清洁 Cu(111)表面上的总吸附态密度(TDOS)以及投影态密度(PDOS)如图 4 所示。显然,在 1/9 ML 处发现了明显的 V 吸附原子的自旋极化(见图 4c),而在 1 ML 处没有发现 V 吸附原子的自旋极化(见图 4f)。此外,在 1/9 ML 和 1 ML 覆盖范围内都没有观察到 Cu 原子的自旋极化(见图 4b、e)。通过对 V 吸附前后 Cu 的 PDOS 积分(即导致 Cu 上的电子数),我们发现 Cu 原子上的电荷略有增加,表明电荷从 V 吸附原子转移到 Cu 衬底V/Cu(111)。换句话说,V 吸附导致对 Cu 的 n 型掺杂。为了更好地理解 Cu(111) 表面上的 V 吸附,我们分别在图 5a、b 中绘制了 1/9 ML 和 1 ML 覆盖的变形电荷密度的等高线图。变形电荷密度定义为 \( \Delta \rho \left(\overrightarrow{r}\right)={\rho}_{\left[V/ Cu(111)\right]}\left(\overrightarrow{ r}\right)-\sum \limits_{\mu =1}^N{\rho}^{atom}\left(\overrightarrow{r}-\overrightarrow{R_{\mu }}\right) \)如图 5 所示,对于 1/9 ML 和 1 ML 覆盖范围,V 吸附原子与其相邻的 Cu 原子之间的共价键和离子键都清晰可见。具体而言,1/9 ML覆盖范围的共价键相对较强(与1 ML相比),而1 ML覆盖范围的离子键相对较强。
<图片>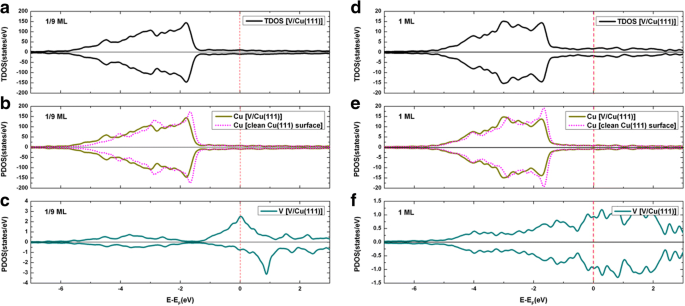
一 V/Cu(111) 系统在 1/9 ML 覆盖率下的 TDOS; b 1/9 ML 时衬底总 Cu 原子的 PDOS; c V adatom 的 PDOS 1/9 ML; d V/Cu(111) 体系在 1 ML 时的 TDOS; e 1 ML 时基板总 Cu 原子的 PDOS;和 f V 吸附原子的 PDOS 为 1 ML。值得注意的是,干净的 Cu(111) 表面的 DOS 也被注入到图中b 和 e 对比
<图片>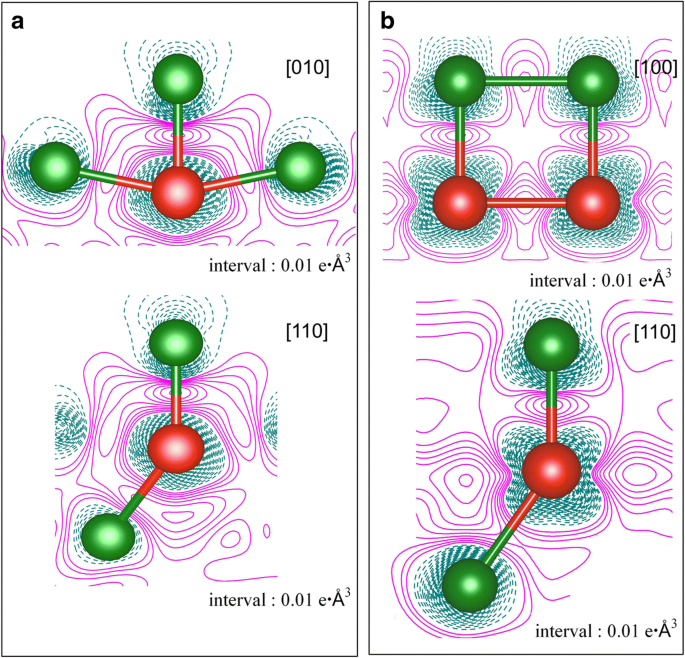
V 原子吸附在 Cu(111) 表面在两个覆盖范围内的变形电荷密度,即 a 1/9 ML 和 b 1 毫升。电子积累和消耗分别由品红色实线和深青色虚线表示。绿色和红色的球分别代表Cu和V原子
Cu(111) 表面的石墨烯层
从上述讨论中的吸附能,发现 subT 位点是 V 原子在清洁 Cu(111) 上最稳定的吸附位点。虽然这样的吸附位点是有兴趣的;然而,留在 Cu 表面层下方的 V 原子可能会导致不利的特性,从而限制其作为表面催化剂的应用。因此,为了减轻 V 吸附原子与 Cu(111) 表面之间的直接相互作用,我们尝试引入缓冲层。由于与 V/Cu(111) 系统最近的晶格匹配,石墨烯是一个完美的选择,这是本工作中首先考虑的。
石墨烯在金属表面的吸附已经在之前的几篇出版物中进行了深入研究 [52,53,54]。当石墨烯吸附在 Cu(111) 上时,考虑了石墨烯/Cu(111) 系统(以下简称 Gra/Cu(111))的三种可能几何形状,即石墨烯位于顶部-fcc、顶部-hcp , 和 fcc-hcp 吸附位点;参见图 6a-c。根据我们的结果,top-fcc 几何结构(图 6a)被证明是能量最稳定的结构,每个碳原子的吸附能为 47 meV,石墨烯和 Cu(111) 表面之间的平衡距离为 3.14 Å,这与之前的研究相当吻合 [52,53,54]。如此低的吸附能 (47 meV/C) 和大的层间距离意味着石墨烯和 Cu(111) 之间的结合相对较弱。图 6d 显示了具有顶部 fcc 配置的石墨烯/Cu(111) 系统的能带结构(图 6a),特别是,石墨烯贡献的电子态在图中由品红色圆圈勾画。独立式石墨烯片的能带结构也显示在图 6e 中以进行比较。从这些图中可以看出,独立片和 Cu(111) 表面的石墨烯的能带结构非常相似。 K 处的狄拉克点(具有线性带交叉)保留在图 6d 中,但当石墨烯吸附在 Cu(111)表面上时有一点下降。交叉点的下降表明电荷从 Cu(111) 衬底转移到石墨烯层,这与之前的结果一致,即 Al、Ag 和 Cu 是由石墨烯掺杂的 n 型 [52,53,54 ]。形变电荷密度,即 Gra/Cu(111) 的总电荷密度与独立石墨烯和清洁 Cu(111) 表面电荷密度之和的电荷差,即 \( \Delta \rho \ left(\overrightarrow{r}\right)={\rho}_{Gra/ Cu(111)}\left(\overrightarrow{r}\right)-{\rho}_{Gra}\left(\overrightarrow{ r}\right)-{\rho}_{Cu(111)}\left(\overrightarrow{r}\right) \),绘制在图 6f 中。如图 6f 所示,我们还可以根据 C 原子周围的实线轮廓线观察电荷从 Cu(111) 衬底到石墨烯层的转移。同时,我们绘制了 Gra/Cu(111) 系统中 C 和 Cu 原子的投影态密度 (PDOS),以及 Cu(111) 表面(有和没有石墨烯吸附)的功函数变化,如图 1 所示。 7. 从PDOS的积分中,我们发现C原子上的电子略有增加,而Cu原子上的电子略有减少,这也验证了电荷转移现象。此外,我们发现在吸附石墨烯层后,Cu(111) 的计算功函数从 4.78 eV 变为 4.68 eV。所有这些都证实了电荷转移是从Cu衬底到石墨烯层。
<图片>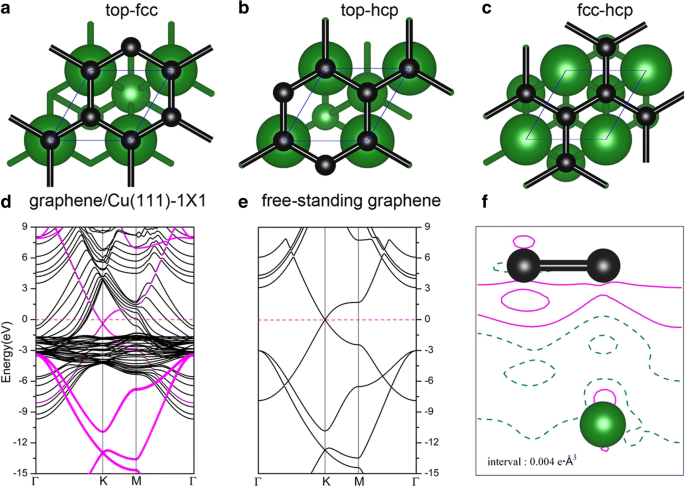
Cu(111)表面石墨烯片的几何形状:a 顶级fcc,b top-hcp 和 c fcc-hcp 站点。 d 与 e 相比,top-fcc 几何结构中石墨烯/Cu(111) 系统的能带结构 那些独立的石墨烯。品红色点表示由石墨烯贡献的电子态。 f 石墨烯/Cu(111) 的总电荷密度与顶部 fcc 几何形状的独立石墨烯和干净的 Cu(111) 表面的电荷密度总和之间的电荷差异。等高线的间隔为 0.004e Å −3
<图片>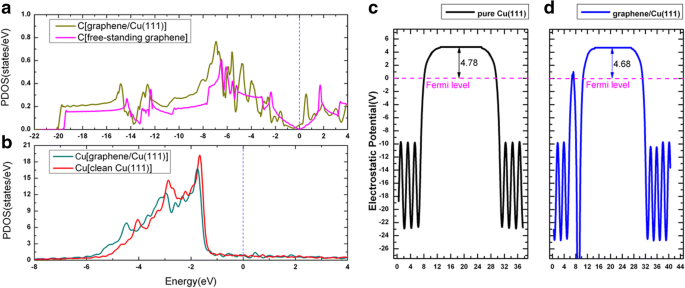
a 的石墨烯/Cu(111) 状态的投影密度 C原子和b 铜原子。 c, d 干净的Cu(111)表面和石墨烯/Cu(111)界面的功函数
钒在石墨烯覆盖的 Cu(111) 表面的吸附
在本节中,我们尝试评估钒在石墨烯覆盖的 Cu(111) 表面上吸附的原子、电子和磁特性。考虑了四种可能的吸附位点,标记为 topA、桥、topB 和空心位点,如图 1d 所示。 V 原子在 1/9 ML 和 1 ML 的 Gra/Cu(111) 上的吸附能分别在图 1e、f 中给出。石墨烯覆盖的 Cu(111) 表面上 V 吸附的能量有利位点是覆盖依赖性的。更具体地说,对于 1/9 ML,V 吸附原子更喜欢最大配位的空心位点(见图 1d),而低配位的顶部位点(即 topA 位点,见图 1d)则更喜欢 1 ML 的高覆盖率. V/Gra/Cu(111)体系的吸附能、V原子与其相邻C原子之间的键长以及钒和碳的原子磁矩列于表1中。 V原子对V原子的吸附能1/9 ML 和 1 ML 的 Gra/Cu(111) 表面 Ead 分别为每 V 原子 1.91 和 1.16 eV,与 Cu(111) 表面上的相比有所降低。显然,石墨烯缓冲层的引入可以减弱 V 吸附原子与 Cu(111) 表面之间的相互作用,正如我们预期的那样。我们进一步研究了不同覆盖率下 V/Gra/Cu(111) 中 V 的铁磁顺序的自旋极化能量。 1/9 ML 的自旋极化能量为 390 meV,而 1 ML 没有自旋极化。与 V/Cu(111) 系统相比,V/Gra/Cu(111) 系统的自旋极化能量要高得多(390 meV 与 110 meV 相比,见表 1)。 V/Gra/Cu(111)体系中V吸附原子在1/9 ML处的磁矩为2.93 μB,接近于3 μB/atom(气相V原子的值),这意味着V原子是隔离良好,V 原子和石墨烯层之间几乎没有电荷转移。还发现了C原子的小磁矩(0.16 μB/atom)。
接下来,我们讨论了石墨烯覆盖的 Cu(111) 表面上 V 吸附的能带结构。图 8 描绘了在 Gra/Cu(111) 表面吸附 V 的能带结构,以及绘制在两个不同晶胞中的独立石墨烯的能带结构。其中,蓝色和红色圆圈分别代表石墨烯和 V 吸附原子的贡献。首先,应该指出的是,有许多铜带穿过费米能级,表明系统中的铜原子对系统的电导有显着贡献。对于 1/9 ML 覆盖,能带结构(见图 8b、c)也表明系统的电子结构是自旋极化的。如上一节所述,对于 Gra/Cu(111),石墨烯与 Cu(111) 表面之间的相互作用非常弱;因此,在整个能带结构中很容易识别出石墨烯贡献的能带。然而,在石墨烯覆盖的 Cu(111) 表面(即 V/Gra/Cu(111) 系统)上吸附 V 后,我们可以看到石墨烯的狄拉克点在自旋向上通道的能带结构,而石墨烯的“线性交叉点”在自旋向下通道中仍然可以区分。尽管如此,自旋下降分量中“线性交叉点”的非常低的偏移表明相对大量的电荷转移到石墨烯层。应该注意的是,转移到石墨烯层的电荷来自 V 原子层(见图 10a),因为 C 和 Cu 层之间的相互作用很弱。对于 1/9 ML 处的自旋通道,我们发现除了大量的 d 基体Cu原子的电子对费米面有贡献,也有很多d V 吸附原子的电子对系统电导的贡献。同时,p的杂交 具有 d 的 C 原子的电子 V 吸附原子和表面 Cu 原子的电子是可见的(但并不显着)。同样,我们在图 8b 中标记了费米面上的两个代表点 (A, B)。 A点代表来自d的贡献 xy , d x 2 − y 2 V 吸附原子的电子,而 B 点显示了 p 杂化的贡献 z 具有 d 的 C 原子的电子 z 2 V 吸附原子的电子和最上层的 Cu 原子。显然,表面V层是重要的导电层。相比之下,在 1/9 ML 的自旋向下通道中,费米能级的电子态主要来源于 d Cu原子和p的电子 z C原子的电子; d 的贡献 V 吸附原子的电子可以忽略不计。对于 1 ML 覆盖范围,图 8e、f 中所示的能带结构意味着该系统是非自旋极化的。由于石墨烯层的狄拉克点也被“破坏”,因此,V吸附原子与石墨烯缓冲层之间的相互作用应该很强。从计算的系统电子态可以看出,对费米能级有贡献的电子主要来自s , d Cu和d的电子 V 原子的电子,以及 p -C 原子的电子。更详细地,位于费米表面上的 k 点 A、B、C、D 和 E 在图 8e 中进行了标记。 A点表示d的杂交 yz 和 d z 2 第一层(最上层)Cu 原子。点 B、C 和 D 是来自 d 的电子态 -V 吸附原子的电子。更具体地说,B 点描述了来自 d 的电子态 xy , d x 2 − y 2 V 吸附原子的电子。此外,E点显示了s的强杂交 , d z 2 具有 p 的 V 吸附原子的电子 z C原子的电子,以及相对较弱的p混合 z 带有 d 的 C 原子的电子 z 2 最上层Cu原子的电子。
<图片>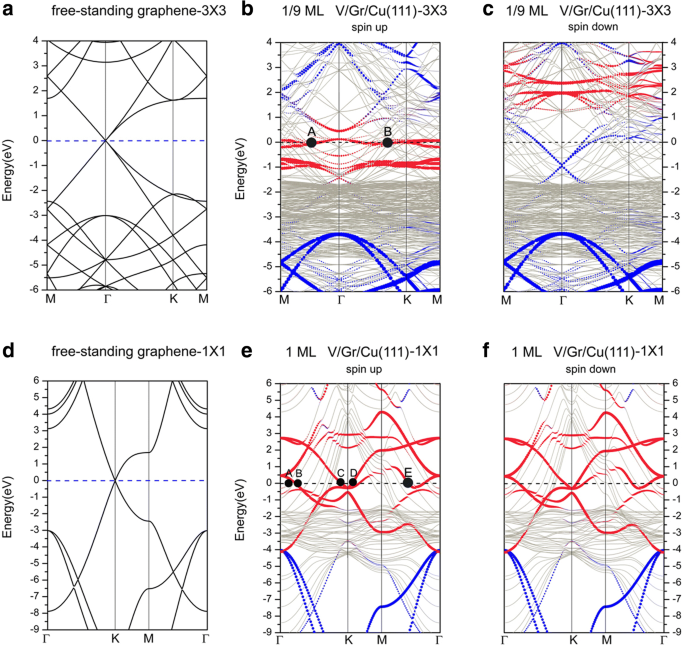
独立石墨烯的能带结构,绘制在 a 的布里渊区 一个 (3 × 3) 晶胞和 d 一个 (1 × 1) 晶胞。 b石墨烯覆盖的Cu(111)表面V吸附的能带结构 旋转 1/9 ML,c 降速 1/9 ML,e 旋转 1 ML,然后 f 降速 1 ML
我们现在介绍 V/Gra/Cu(111) 系统的态密度。对于 1/9 ML 和 1 ML 覆盖,石墨烯覆盖的 Cu(111) 表面上 V 吸附的总状态密度以及状态的投影密度如图 9 所示。 At 1/9 ML, the spin polarizations of V adatoms and C atoms in the graphene layer are clearly seen (see Fig. 9c, d), while no spin polarization is found for Cu atom (see Fig. 9b). At 1 ML coverage, no spin polarization has been found for all atoms (see Fig. 9f-h). At 1/9 ML coverage, the DOS of the spin-up channel at the Fermi level is mainly contributed from the Cu atoms (totally 11.9 states/eV∙u.c.) and V atoms (totally 5.8 states/eV∙u.c.), with only minor contributions from the graphene layer (totally 0.4 states/eV∙u.c.). Meanwhile, the DOS of spin-down channel at the Fermi level is mainly contributed from the Cu atoms (totally 11.9 states/eV∙u.c.) and graphene layer (totally 1.1 states/eV∙u.c.), with only minor contributions from the V atoms (totally 0.1 states/eV∙u.c.). For the 1 ML coverage, both the DOS of spin-up and spin-down channels at the Fermi level are mainly contributed from the Cu atoms and V atoms (i.e., 1.1 and 0.7 states/eV∙u.c. for each spin component, respectively), with negligible contribution from the graphene layer (0.04 states/eV∙u.c). By integrating the PDOSs for each atom before and after the V adsorptions (leading to number of electrons), the charge transfer can be determined for different atoms. To be specific, the total valence electrons of the Cu atoms are reduced slightly for both the 1/9 ML and 1 ML coverages when compared with those of a clean Cu(111) surface, while the total valence electrons of C atoms are slightly increased when compared with that of a free-standing graphene. This implies that small amount of charges are transferred from Cu substrate to graphene layer for V/graphene/Cu(111) systems regardless of the V coverages. The total valence electrons of Cu atoms in V/Gra/Cu(111) systems are almost equal to those in the graphene/Cu(111) systems, which indicates that the Cu substrate has not been affected by V adsorption. The physical pictures given by the analysis of DOSs here are all in consistent with the analysis of the band structures. Finally, we show in Fig. 10 the contour plots of the deformation charge densities for the 1/9 ML and 1 ML coverages, respectively. The deformation charge density is defined as \( \Delta \rho \left(\overrightarrow{r}\right)={\rho}_{\left[V/ Gra/ Cu(111)\right]}\left(\overrightarrow{r}\right)-\sum \limits_{\mu =1}^N{\rho}^{atom}\left(\overrightarrow{r}-\overrightarrow{R_{\mu }}\right) \). As shown in Fig. 10, the interactions between the graphene layers and the substrate Cu atoms are both relatively weak for 1/9 ML and 1 ML coverages, which are in consistent with the above discussions. From Fig. 10a, for the 1/9 ML, the bonding between V adatoms and its adjacent C atoms is mainly ionic, and the covalent bonding is not obvious. In contrast, for the 1 ML coverage, both ionic and covalent bonding between V adatom and its adjacent C atoms are clearly visible (see Fig. 10b). Besides, the covalent bonding between neighboring V adatoms is also very significant at 1 ML coverage. Due to the existence of graphene buffer layer, V adatoms cannot interact directly with the Cu atoms.

一 TDOS of V adsorption on graphene-covered Cu(111) surface at 1/9 ML coverage; PDOS for b Cu atoms, c C atoms, and d V adatoms at 1/9 ML. Likewise, e TDOS of V adsorption on Gra/Cu(111) surface at 1 ML coverage; PDOS for f Cu atoms, g C atoms, and h V adatoms at 1 ML. For comparison, PDOS of Cu atoms of a clean Cu(111) and graphene/Cu(111) are implanted in b 和 f , while PDOS of C atoms of a free-standing graphene are also implanted in c 和 g

Deformation charge densities for the adsorption of V atoms on the graphene-covered Cu(111) surface at two coverages, i.e., a 1/9 ML and b 1 ML. Electron accumulation and depletion are represented by magenta solid lines and green dashed lines, respectively. The green, black, and red balls represent Cu, C, and V atoms, respectively
We have also calculated the phonon spectra for both the V/Cu(111) and V/Gra/Cu(111) systems. From the calculated phonon spectra, we find that there is no “imaginary frequency” for both the two types of systems, indicating that the systems studied are dynamically stable and would be seen in the laboratory. Since the main purpose of our work is not the thermodynamic stability, therefore the figures of the phonon dispersions are not shown in this text. Second, we have noticed that the different DFT functionals we adopted may lead to the different results. Hence, we have calculated the 1 ML V/Gra/Cu(111) system (as a representative) within the DFT framework under the B3LYP, HSE06 hybrid functionals, as well as the PBE functional. The results suggest that the adsorption site with largest adsorption energy is the topA site, calculated from all the PBE, HSE06, and B3LYP methods. However, relative adsorption energies at different adsorption sites from the B3LYP and PBE and HSE06 methods differ significantly (results from PBE and HSE06 methods are almost the same, since this is a metallic system). On the other hand, the geometrical parameters obtained from the three functionals show good consistency. Although the detailed charge density contours are somewhat different between PBE and B3LYP method, the main bonding characteristics are the same from both the two methods. In summary, the main point is that the adsorption energies obtained from B3LYP functional are significantly larger than those from the PBE and HSE06 functionals. To explain this point, Paier et al. argued that B3LYP functional lacked of a proper description of the “free-electron-like” systems with a significant itinerant character (e.g., metals and small gap semiconductors). They have concluded that the overestimation of the total energy of the atoms can be induced by the significantly overestimation on the exchange and correlation energies of B3LYP functional. In this respect, PBE functional often shows much more reliable results [55].
结论
To summarize, using first-principles calculations, we have systematically investigated the electronic and geometric properties of the adsorption of V atoms on both the clean Cu(111) surface and the graphene-covered Cu(111) surface. Firstly, for the V/Cu(111) system, an adsorption site underneath the Cu surface layer is found as the preferable adsorption site for V atom regardless of the coverages. The hybridization of V’s d states with Cu’s d states rules the electronic properties of V/Cu(111) systems. Ferromagnetic order of V adatoms is energetically favored for 1/9 ML coverage (1.34 μB/atom), while no magnetism of V adatoms is observed for 1 ML coverage. Due to the strong interaction between V adatom and its adjacent substrate’s Cu atoms, the magnetic moment of V is significantly reduced. Secondly, the graphene/Cu(111) systems are investigated and the results agree well with the previous literatures. Thirdly, adsorptions of V on the graphene-covered Cu(111) at two coverages (i.e., 1/9 ML and 1 ML) show different preference of adsorption sites. The hollow site with maximum coordination is energetically favored for the adsorption of 1/9 ML, while the top site with low coordination is preferred for 1 ML adsorption. In V/Gra/Cu(111) systems, the interactions of C atoms with the V adatoms destroy the electronic properties of both the original graphene layer and the adsorbed atoms, represented by the strong hybridization of C’s p z -states with V adatoms’ d z 2 -states. A net magnetic moment for C atoms of graphene also appeared (0.16 μB/per carbon). In short, our study paves the way to a deep understanding of the adsorption properties of vanadium atoms on the clean Cu(111) and graphene-covered Cu(111) substrates. Simultaneously, this study also provides a reference for the possible applications of the V/Cu(111) and V/Gra/Cu(111) systems in the catalyst in nanomaterials industry, spintronic devices, and others.
缩写
- Cu(111):
-
(111) surface of copper
- FM:
-
Ferromagnetic
- GGA:
-
Generalized gradient approximation
- ML:
-
单层
- PAW:
-
Projector augmented wave
- PBE:
-
Perdew-Burke-Ernzerhof
- V/Cu(111):
-
Vanadium atoms adsorbed on Cu(111) surface
- V/Gra/Cu(111):
-
Vanadium atoms adsorbed on graphene-covered Cu(111) surface
- VASP:
-
Vienna ab initio simulation package
- vdWs:
-
van der Waals interactions
纳米材料
- 钴掺杂 FeMn2O4 尖晶石纳米粒子的制备和磁性
- 走向 TiO2 纳米流体——第 1 部分:制备和性质
- 过渡金属掺杂高岭石纳米粘土的结构和电子特性
- 垂直电场对 ML-GaS 的电子和光学各向异性特性的调制
- 基于苯基三甲氧基硅烷改性氧化铝纳米颗粒的 Al2O3:SiOC 纳米复合材料的形成和发光特性
- 通过界面层设计调整 ZnO 薄膜的表面形貌和特性
- 飞秒激光诱导硫超掺杂硅 N+/P 光电二极管的光学和电子特性
- 探测 Ag n V (n =1-12) 簇的结构、电子和磁特性
- InSe 纳米带的电子结构和 I-V 特性
- Pr2CuO4 纳米片的可控合成和选择性吸附特性:机理讨论
- GaTe/C2N 异质结构中的应变可调电子特性和能带排列:第一性原理计算
- 汽车 PCB 特性和设计注意事项