

掺氮多孔碳纳米片与 Mo2C 纳米颗粒强耦合,可实现高效电催化析氢
摘要
探索用于水电解的地球丰富且不含贵金属的催化剂对于可再生氢生产至关重要。在此,通过一种新方法合成了一种高活性的氮掺杂多孔碳纳米片与 Mo2C 纳米颗粒偶联的电催化剂(Mo2C/NPC),BET 表面积高达 1380 m 2 g −1 使用KOH活化碳复合材料。 KOH 在蚀刻 MoS2 以产生 Mo 前驱体的过程中起关键作用;同时,它腐蚀碳形成多孔结构并产生还原性气体如 H2 和 CO。所得的 Mo2C/NPC 杂化物在酸性溶液中表现出优异的 HER 活性,在 10 mA cm − 的电流密度下具有 166 mV 的过电位2 , 起始过电位为 93 mV,塔菲尔斜率为 68 mV dec −1 ,以及卓越的长期循环稳定性。该策略为制备其他用于能量转换和存储的金属碳化物/碳杂化物提供了一种有前景的策略。
介绍
当今,环境污染和能源危机已成为可持续发展的关键问题[1, 2]。解决问题的关键是实现清洁的可再生能源。通过催化剂分解水产生的氢气被认为是化石燃料的有前途的替代品 [3, 4]。铂基催化剂仍然是迄今为止最有效的析氢反应(HER)催化剂,但其稀缺性和高成本限制了其大规模应用。因此,过渡金属硫化物 [5]、氧化物 [6]、氮化物 [7]、磷化物 [8, 9] 和碳化物 [10] 等低成本且地球资源丰富的过渡金属化合物出现作为贵金属的替代品。金属催化剂。在这些电催化剂中,Mo2C 作为一种高效的 HER 电催化剂引起了极大的兴趣,因为它的 d 带电子结构类似于 Pt [11]。碳化钼的催化性能主要依赖于更多活性位点的暴露和提高催化剂的导电性。研究人员倾向于改进Mo2C杂化物的组成和结构;然而,目前Mo2C杂化物的合成主要需要高温,这会导致颗粒团聚,导致活性表面减少,降低HER催化性能[12]。为了减少碳化钼的团聚,通常使用导电碳支撑材料来增加活性表面和导电性。具有二维结构的石墨被证明是一种极好的催化剂载体材料[13]。否则,催化剂的高表面积会提供更多的活性位点暴露,从而提高 HER 性能。不幸的是,最近提高催化剂比表面积的方法仍然有限,研究人员倾向于努力减小催化剂的尺寸,很少关注增加材料的孔隙率[14, 15]。因此,限制了 Mo2C/C 复合材料比表面积的增加。高比表面积(4196 m 2 )多孔炭的制备 g −1 ) 来自氢氧化钾活化聚合物水凝胶 [16] 为合成具有多孔结构的支撑导电石墨基材提供了新思路,该多孔结构将为 HER 过程中的反应物提供开放空间和短扩散通道 [17]。先前的报告已经证明,碳材料中 Mo2C 和 N 掺杂剂之间的协同效应将导致高 HER 电催化性能 [18]。作为支撑基材的 N 掺杂多孔碳纳米片的受控合成将具有高表面积、优异的导电性、高耐久性、N 掺杂剂以增强电子转移和多孔结构以促进质量/电荷传输。此外,有报道证明具有六方结构的 β-Mo2C 是碳化钼四相中最活跃的相,因为它具有类似于 Pt 的价带形状 [19]。因此,合成掺氮多孔碳纳米片与β-Mo2C纳米颗粒用于高效催化制氢是一个挑战。
在此,我们报告了一种新的自模板方法,以实现具有高孔隙率的高活性和稳定的无贵金属电催化剂。商业 MoS2 用作 Mo 源,自模板和多巴胺分别用作 C 和 N 源。由于多巴胺可以很容易地在钼源表面自聚合形成聚多巴胺(PDA)微球,因此合成具有更多活性表面暴露在空气中的催化剂至关重要[20]。报道者倾向于使用 SiO2 [21] 和 NaCl [22] 等模板来避免聚集并形成具有高比表面积的结构。然而,溶解二氧化硅需要氢氟酸,这是一种高风险化学品,去除盐模板涉及更多步骤。我们选择商业 MoS2 作为 Mo 源和自模板,因为 MoS2 可以在高温下与 KOH 反应。模板的去除和 KOH 的活化导致多孔碳和还原气体合成了具有高催化活性的最终 Mo2C/NPC 杂化物。我们的合成方法为制备不含贵金属的高性能HER催化剂提供了一种很有前景的策略。
方法
Mo2C/NPC Hybrid 和参考NPC 的制备
在典型的合成中,首先通过超声处理将 500 mg 商业 MoS2 分散在 100 ml 去离子水中。然后,将 120 mg Trizma® 碱和 200 mg 多巴胺盐酸盐加入悬浮液中。混合物在室温下搅拌24 h,用去离子水洗涤后过滤收集产物。将其放入烘箱中过夜后,将得到的 MoS2@PDA 在管式炉中于 600 °C 下碳化 2 h 以形成 MoS2@NC。将碳化的 MoS2@NC 浸泡在 4 ml 的 7 M KOH 中,KOH 与 MoS2@NC 的质量比为 3:1。干燥的 KOH/MoS2@NC 混合物在 N2 下在 800 °C 下加热 1 小时。冷却后,将样品过滤并用稀盐酸溶液和去离子水洗涤。然后在 60 °C 下干燥过夜。最终产物为Mo2C/NPC,除不添加商业化MoS2外,按照类似步骤得到N掺杂多孔碳(NPC)。
特征化
使用 Cu Kα 辐射 (λ =1.54056 Å)。使用场发射扫描电子显微镜(SEM,Hitachi SU8020)表征形态。使用 FEI Tecnai G2 F20 S-TWIN TMP 进行透射电子显微镜 (TEM) 图像和相应的能量色散 X 射线 (EDX) 元素映射图像。拉曼光谱用共焦拉曼光谱仪(HORIBA LabRAM HR Evolution)记录。 X射线光电子能谱(XPS)在PHI Quantera-II扫描X射线微探针光谱仪上进行,以Al Kα辐射(1486.6 eV)作为激发源。 TGA/DSC 曲线由 TGA/DSC1 Mettler-Toledo 热分析仪测量。样品的比表面积用Micromeritices ASAP 2020 HD88测定。
电化学测量
所有电化学测试均在CHI660E恒电位仪(CH仪器,中国)上用标准三电极系统进行,本文所有电位均指可逆氢电极(RHE),根据E(RHE) =E(Ag/AgCl ) + 0.059 pH + 0.197 V. 石墨棒用作对电极,Ag/AgCl(饱和氯化钾填充)分别用作参比电极。直径为5 mm的玻碳电极被15 μL催化剂墨水覆盖,用作工作电极。通常,在制备工作电极时,将 4 mg Mo2C/NPC 和 20 μL Nafion 溶液通过超声处理 1 h 分散在 1 mL 3:1 v/v 水/乙醇中,形成均质墨水。电化学测试前,将新鲜的工作电极循环50次以稳定电流,并在0.5 M H2SO4中以5 mV s -1 的扫描速率测试线性扫描伏安法(LSV) 没有红外补偿。此外,循环伏安图 (CV) 从 0 到 0.2 V(相对于 RHE,在 0.5 M H2SO4 中)获得,扫描速率为 20、40、60、80、100、120 和 140 mV s −1上> , 分别。
结果和讨论
Mo2C/NPC 杂化物的合成过程如图 1 所示。我们选择多巴胺作为碳源和氮源。选择商业块状 MoS2 作为 Mo 源和自模板,其尺寸为 ~ 2 μm(附加文件 1:图 S1a)。首先,多巴胺在块状 MoS2 表面自聚合形成 MoS2@PDA 核壳结构(附加文件 1:图 S1b)。然后,核壳结构 MoS2@PDA 被碳化以形成包裹在 MoS2 表面的 N 掺杂碳膜,标记为 MoS2@NC(附加文件 1:图 S1c)[23, 24]。最后,将制备的 MoS2@NC 和 KOH 的混合物放入管式炉中并反应以获得最终产品:掺氮多孔碳纳米片与 Mo2C 纳米颗粒(作为 Mo2C/NPC 捐赠)(附加文件 1:图 S1d)。当MoS2作为Mo源被切断时,多巴胺在MoS2表面形成PDA薄膜,MoS2作为自模板避免多巴胺形成微球,生成PDA薄膜。这是因为从 PDA 到 N 掺杂的 C 的转换将继续保持其形态 [15];当 MoS2 与 KOH 反应时,我们可以得到长度约为 2 μm 的碳纳米片。 MoS2@NC 中的碳也可以通过 KOH 活化得到多孔 C 纳米片。 Mo2C/NPC 的形成可以基于一系列反应提出。 KOH插入并与碳反应的过程可以概括为KOH活化反应,化学反应方程式描述为6KOH+2C↔2K+3H2+2K2CO3,K2CO3可以进一步分解为K2O、CO2和CO[25 ]。 KOH活化反应的过程不仅能腐蚀碳单元,产生碳的多孔结构,还能促进石墨碳的形成。同时,KOH 可以通过硫蒸气的扩散和 K2S 的形成来蚀刻 MoS2 模板以产生 Mo2C 纳米颗粒。因此,反应导致形成Mo2C/NPC杂化物。
<图片>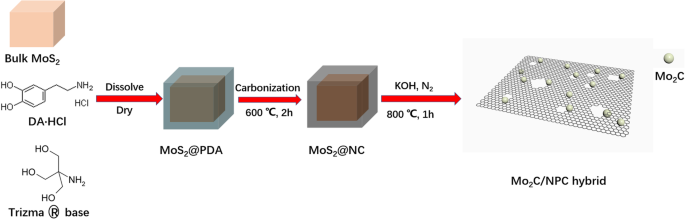
Mo2C/NPC杂化物制备流程示意图
通过 X 射线衍射检查产物的晶相组成(图 2a)。 26°附近的宽峰和46.3°处的峰可归因于石墨碳的(003)和(012)面。 34.3、37.9、39.39、52.1、61.5、69.5、74.6和75.5°处的其他X射线衍射峰归因于(100)、(002)、(101)、(102)、(110)的衍射)、(103)、(112) 和 (201) 六方 β-Mo2C 面(JCPDS 35-0708)。此外,没有可辨别的杂质,如钼金属、氧化物、硫化物或其他碳化物,表明商业 MoS2 完全转化为 Mo2C。图 2b 中的拉曼光谱结果进一步证实,所制备的催化剂是碳化钼和石墨的混合物。 G带与D带的强度比,I G/我 D> 1,表明碳基本上是石墨[26]。根据空气中的热重分析 (TGA),发现最终产品中 Mo2C 的量为 ~ 44 wt%(附加文件 1:图 S2)。在 77 K 下测量氮吸附-解吸等温线以评估 Brunauer-Emmett-Teller (BET) 比表面积。如图 2c 所示,Mo2C/NPC 的氮吸附-解吸等温线呈现 H4 型滞后回线,适用于具有微细孔的材料。此外,BJH解吸平均孔径计算为3.23 nm,BET比表面积为1380 m 2 g −1 ,这揭示了多孔结构的成功合成。这种具有超高表面积的碳基微孔结构被认为是一种理想的电极材料,它不仅可以为反应物提供开放空间和短扩散通道,还可以促进H + 和H2的解吸,从而导致良好的质/电荷转移能力。
<图片>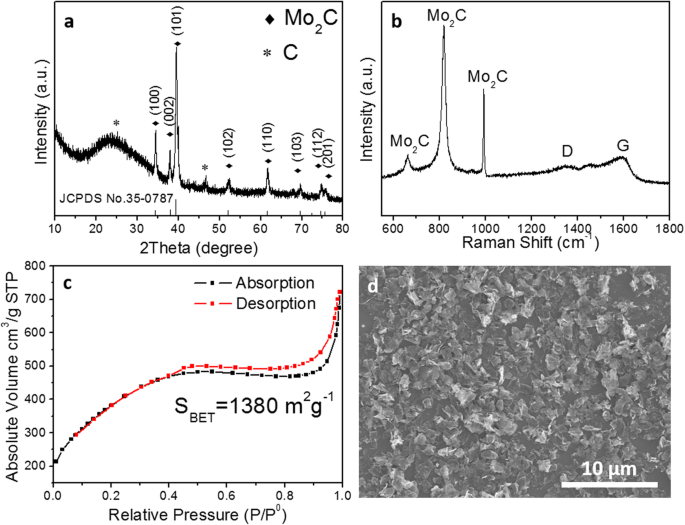
Mo2C/NPC 的物理特性。 一 XRD 图,b 拉曼光谱,c N2 吸附-解吸等温线和d SEM图像
然后,通过SEM和TEM研究了分层Mo2C/NPC杂化物的形态和结构。如图 2d 所示,低倍 SEM 图像呈现出许多分散良好的纳米片状结构,平均尺寸为 2 μm,与模板 MoS2 的尺寸一致。图 3a 和 c 中的 TEM 图像显示,尺寸从几纳米到 50 nm 的 β-Mo2C 纳米颗粒锚定在氮掺杂的碳纳米片上。从图 3b [27] 中的 TEM 图像可以看出碳纳米片的多孔性质。此外,图 3d 中的高分辨率 TEM 图像显示了 d 间距为 0.23 nm 和 0.24 nm 的晶格条纹,它们对应于 β-Mo2C 的(101)和(002)平面。负载碳的多孔结构以及 Mo2C 纳米粒子与 N 掺杂的多孔 C 纳米片的耦合将促进电子从碳化钼转移到碳,从而提高催化剂的效率。如图 3e 所示,能量色散谱 (EDS) 分析表明纳米片由 Mo、C 和 N 元素组成,证实了 Mo2C/NPC 杂化物的成功合成。
<图片>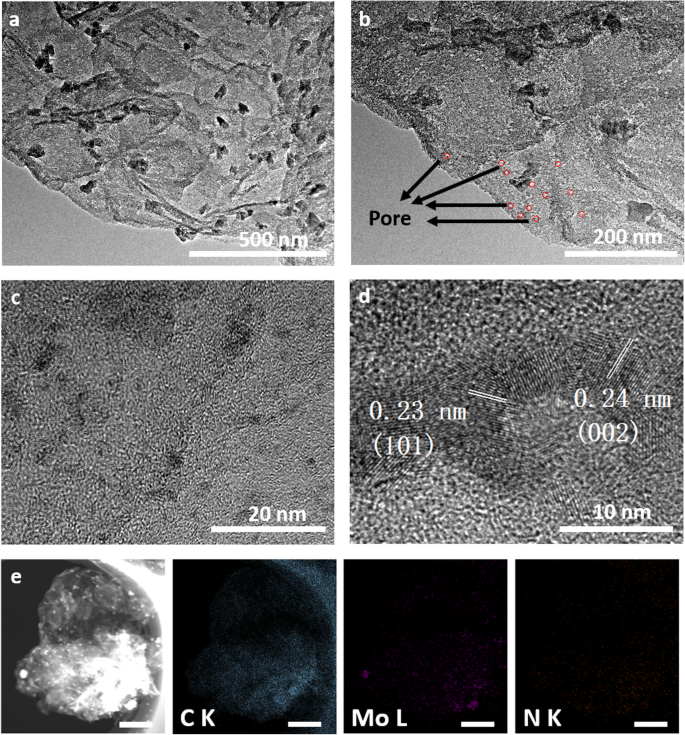
Mo2C/NPC 的形貌。 一 –d 不同放大倍数和 e 下的 TEM 和 HRTEM 图像 Mo2C/NPC对应的EDS元素映射(比例尺500 nm)
XPS 进一步阐明了合成的 Mo2C/NPC 纳米片的表面组成。从图 4a 中显示的调查光谱中,可以清楚地识别 Mo、C、N 和 O 元素。 C 1s XPS 峰可以拟合成三个以 284.6、285.6 和 288.8 eV 为中心的峰(图 4b),这可以分别归因于 CC/C=C、CN 和 C=O 物种 [28, 29 ]。 Mo 3d XPS 峰可以解卷积成两个双峰(图 4c)。一个以 228.6/231.6 eV 的结合能为中心,另一个以 232.9/235.9 eV 为中心,这可以分别归因于 Mo2C 和表面氧化的 MoO3[14,26,29]。大量氧化钼不可避免地大量存在,这是由于碳化钼暴露在空气中时,其表面缓慢氧化 [30]。此外,据报道,在碳化物表面形成的氧化物可以保留碳化物的活性。结合能分别为 398.4、400.2 和 401.4 eV 的 N 1s 峰(图 4d)可分别归因于吡啶、吡咯和季 N 原子 [24, 29]。先前的报道已经证明碳中的N掺杂剂可以诱导电子转移过程(Mo2C→C→N),从而增强碳中Mo2C和N掺杂剂之间的协同作用[18]。
<图片>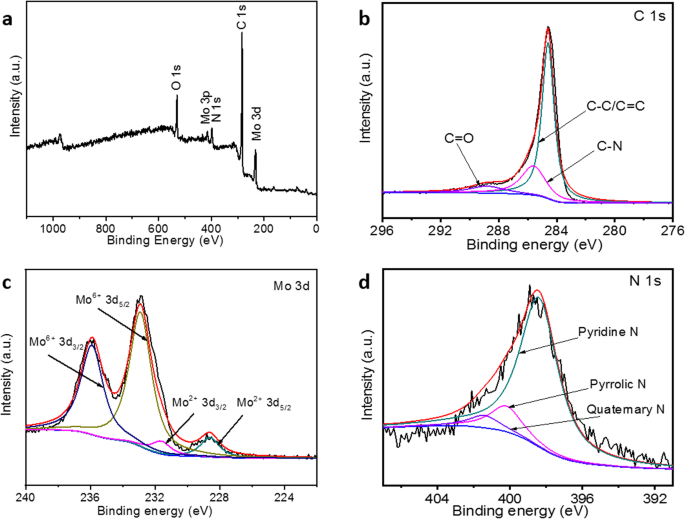
XPS 调查频谱 (a ) 和 C1 的高分辨率 XPS 扫描 (b ), Mo3d (c ) 和 N1 s (d ) Mo2C/NPC
Mo2C/NPC 的电催化 HER 活性首先在 0.5 M H2SO4 中进行评估。为了比较,还使用相同的负载量测试了原始商业 MoS2 (c-MoS2)、N 掺杂多孔 C (NPC) 和 20 wt% Pt/C。图 5a 比较了相应的极化曲线。正如预期的那样,NPC 和商业 MoS2 都显示出非常有限的 HER 活性,起始过电位分别为 354 mV 和 289 mV,而 Mo2C/NPC 的起始过电位为 93 mV,远低于 NPC 和 c-二硫化钼。 Mo2C/NPC 在电流密度为 10 mA cm −2 下的过电位 为 166 mV,远低于 NPC 和原始 c-MoS2,并且与其他工作中的 Mo2C/C 杂化物相当 [20, 31]。为了探索催化剂的 HER 动力学,将 Tafel 图拟合到 Tafel 方程 (η =a + b 日志 (j )),其中 b 是塔菲尔斜率。如图 5b 所示,Mo2C/NPC 的 Tafel 斜率计算为 68 mV dec -1 , 远低于 c-MoS2 (184 mV dec −1 ) 和 NPC (296 mV dec −1 ),表明解吸步骤在 Mo2C/NPC 催化剂的表面上是有效的。 Mo2C/NPC 混合物的 Tafel 斜率落在 40–120 mV dec −1 ,这意味着在 Mo2C/NPC 表面发生的 HER 遵循 Volmer-Heyrovsky 机制 [32]。根据 Tafel 分析,交换电流密度 (j 0) Mo2C/NPC 的计算结果为 37.4 μA cm -2 ,其性能优于文献中报道的许多非贵重 HER 电催化剂(如附加文件 1:表 S1 中所示)[33,34,35]。为了估计工作条件下 Mo2C/NPC 的电化学活性表面积 (ECSA),我们计算了双层电容 (C dl) 来自图 5c 中不同扫描速率下的循环伏安法 (CV) 曲线。如图 5c 的插图所示,0.1 V 处的电流密度与扫描速率的线性相关表明 C Mo2C/NPC 的 dl 为 102.4 mF cm −2 .如果我们假设标准值为 60 μF/cm 2 ,Mo2C/NPC的ECSA估计为∼ 558 m 2 /g(计算结果见附加文件 1:图 S3)。如此高的 ECSA 来自 Mo2C 和碳载体。由于碳更轻,估计 N 掺杂的多孔 C 占 ECSA 的大部分 [26] 并且它与比 BET 表面积一致,因此支持大多数活性 Mo2C 表面是电化学可及的。
<图片>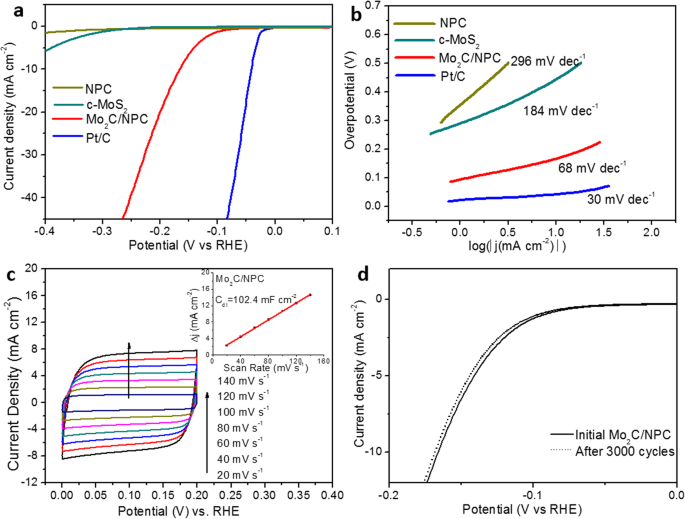
Mo2C/NPC 杂化物在 0.5 M H2SO4 中用于 HER 电催化的电化学测量。 一 极化曲线和b Mo2C/NPC 的 Tafel 图与 Pt/C 基准、c-MoS2 和 NPC 的比较。 c Mo2C/NPC 在 20 到 140 mV/s 不同扫描速率下的 CV 曲线。插图说明了 0.1 V 时的电容电流与扫描速率的关系图。 d Mo2C/NPC在3000个电位循环前后的极化曲线
除了 HER 活性外,稳定性是评估催化剂的另一个决定性因素。进行长期循环伏安法以测量 Mo2C/NPC 在 0.5 M H2SO4 中的稳定性。图 5d 中 Mo2C/NPC 的 HER 极化曲线显示在 3000 次循环后仅 2 mV 损失,表明催化剂的稳定性可以忽略不计。 Mo2C/NPC 在 - 0.166 V 与 RHE 的过电位下的计时电流响应曲线在附加文件 1:图 S4 中说明。基于上述电化学研究,Mo2C/NPC纳米片优异的电催化性能可归因于以下因素:(1)催化剂的高比表面积会导致更多的H + 吸收,支撑基板良好的导电性会提高电子传输; (2) β-Mo2C 纳米粒子与 N 掺杂的多孔 C 纳米片的偶联将扩大催化剂与电解质的接触,促进电荷和传质; (3) 掺杂的 N 原子不仅可以更好地与 H + 相互作用 与C原子相比,还改变了相邻Mo和C原子的电子结构,使Mo2C/NPC成为一种高效的催化剂。
结论
总之,通过 KOH 活化方法开发了一种制备分层 Mo2C/NPC 杂化物的新策略。商业 MoS2 用作 Mo 源和自模板,而多巴胺用作 C 和 N 源。 MoS2 被 KOH 蚀刻产生 Mo 前驱体,碳化的 PDA 被 KOH 腐蚀形成多孔石墨基板。 Mo2C/NPC杂化物优异的HER活性,在10 mA cm −2 时的过电位为166 mV , 起始过电位为 93 mV, Tafel 斜率为 68 mV dec −1 和出色的长期循环稳定性归因于氮掺杂含量、多孔导电基材、丰富的活性位点以及 Mo2C 与石墨碳之间的强相互作用。这种有效的方法可用于设计和制备其他具有高比表面积的碳化物化合物,用于各种电催化应用。
数据和材料的可用性
所有数据完全可用,不受限制。
缩写
- c-MoS2:
-
商用二硫化钼
- 她:
-
析氢反应
- HRTEM:
-
高分辨透射电子显微镜
- Mo2C/NPC:
-
氮掺杂多孔碳纳米片与Mo2C纳米颗粒的偶联
- MoS2@NC:
-
包裹在MoS2表面的掺氮碳膜
- NPC:
-
掺氮多孔碳
- PDA:
-
聚多巴胺
- Pt/C:
-
铂/碳催化剂
- RHE:
-
可逆氢电极
- TGA:
-
热重分析
纳米材料
- 具有可控厚度的二硫化钼用于电催化析氢
- S、N 共掺杂石墨烯量子点/TiO2 复合材料用于高效光催化制氢
- 用贵金属纳米粒子装饰的电纺聚合物纳米纤维用于化学传感
- 碳纳米点作为双模式纳米传感器用于选择性检测过氧化氢
- 具有分层多孔结构的单分散碳纳米球作为超级电容器的电极材料
- 探索 Zr-金属-有机框架作为制氢的高效光催化剂
- 用于高效光催化制氢的 ZnO@TiO2 空心球的分级异质结构
- PtNi 合金助催化剂改性曙红 Y 敏化 g-C3N4/GO 杂化物以实现有效的可见光光催化产氢
- 通过将 Cd0.5Zn0.5S QD 加载到 Ni2P 多孔纳米片上来增强光催化产氢
- 带有 Ag 纳米颗粒的对齐良好的 TiO2 纳米管阵列,用于高效检测 Fe3+ 离子
- 具有部分表面改性的 ZnO 多孔纳米片,可在太阳辐射下增强电荷分离和高光催化活性
- 用于锂/硫电池的 TiO2/多孔碳复合装饰隔板