

Ru/SnxTi1−xO2 柴油氧化催化剂的新型一步水热制备及其低温性能
摘要
金红石Snx Ti1−x 氧气 (x =0, 0.33, 0.5, 0.67, 1) 以钛酸四丁酯和五水氯化锡(IV)为原料,采用一步法水热法合成固溶体。一系列 Ru/Snx Ti1−x 然后通过在 RuCl3 中的浸渍过程制备 O2,以研究 CO 和 C3H8 氧化的性能和稳定性。通过XRD、N2吸附-解吸、FT-IR、TEM、XPS、H2-TPR和O2-TPD技术对这些催化剂进行了表征。 Sn/Ti摩尔比和水热条件对Ru/Snx低温催化氧化性能及稳定性的影响 Ti1−x O2进行了调查。结果表明,Ru/Sn0.67Ti0.33O2催化剂在低温下表现出优异的活性和稳定性。 CO 转化率在 180 °C 时达到 50%,在 240 °C 时达到 90%。此外,C3H8在320℃时转化率达到50%,C3H8在500℃时完全转化,催化反应12 h后没有失活。 Ru/Sn0.67Ti0.33O2 优异的低温活性和稳定性归因于以下因素。首先,XRD 结果表明 Sn 4+ 成功引入TiO2晶格中替代Ti 4+ 形成均匀的固溶体(含有–Sn 4+ –O–Ti 4+ – 物种),这与 TEM 和 N2 吸附-解吸结果一致。 Sn的引入可以抑制锐钛矿晶体的生长,促进金红石相的形成,这种相变有助于提高催化剂的低温活性。其次,TEM 图像显示超细 Ru 纳米粒子(~5 nm)分散在 Sn0.67Ti0.33O2 载体上,表明形成了 Snx Ti1−x O2固溶有利于Ru颗粒的分散。
背景
柴油机由于具有油耗低、热效率高、稳定性好等优点,广泛应用于交通、矿山、工程机械等领域[1]。然而,一氧化碳 (CO)、未燃烧的碳氢化合物 (HC)、各种氮氧化物 (NOx )、柴油车尾气中的颗粒物(PM)对生态环境和人类健康造成严重威胁[2, 3]。此外,严格的环境法律和法规推动了柴油排放控制技术的最新进展。由柴油氧化催化剂(DOC)、选择性催化还原(SCR)和催化柴油微粒过滤器(DPF)组成的一体化尾气后处理系统已被广泛用于净化柴油机尾气。 DOC在后处理系统中的作用是将CO、HCs和NO转化为CO2、H2O和NO2,NO2作为后续脱NOx的原料 反应促进 SCR 反应。此外,它还可以氧化可溶性有机成分 (SOF) 以减少 PM 排放。柴油车冷启动时HCs燃烧不完全,会导致HCs超标排放。因此,催化剂需要在低温下快速点燃[4]。目前,负载在碳材料或氧化物(如 TiO2、Al2O3、CeO2 和 ZrO2)上的贵金属催化剂(如 Pt、Pd 和 Rh)是商业化的柴油氧化催化剂,具有良好的 CO、NO 和 HCs 催化氧化性能.然而,商业化催化剂存在热稳定性差、CO自抑制能力强、成本高等缺点[5]。
Ru和RuOx 催化剂广泛应用于氧化 CO [6]、甲烷 [7] 和氯苯 [8]。重要的是,Ru 催化剂具有出色的低温活性和抗毒性 [8,9,10,11]。但是 Ru 和 RuOx 容易烧结,导致活性位点的暴露减少[12]。因此,Ru催化剂应负载在载体上以防止其烧结并提高催化活性。
TiO2 已被广泛用于净化柴油机尾气。 RuOx 和金红石相TiO2具有相似的晶格常数,Ru/TiO2催化剂中的金红石TiO2在稳定RuOx中起重要作用 与锐钛矿负载的 RuOx 相比,煅烧过程中的颗粒 催化剂。因此,RuOx 可以高度分散在 TiO2 表面。此外,RuOx 之间存在协同效应 和 TiO2,有利于提高 Ru/TiO2 的氧化还原能力 [13,14,15,16,17,18]。为了进一步提高热稳定性、活性成分的分散性以及锐钛矿向金红石相的转化,许多研究引入了Sn 4+ 进入 TiO2 形成 Snx Ti1−x O2 固溶体。黄等人。 [16]发现引入Sn 4+ 进入 TiO2 晶格可以提高 CuO/Tix 的稳定性 Sn1−x O2催化剂和CuO的分散。 Bai等[17]指出Sn 4+ 显着提高了 TiO2 的热稳定性。梅赫拉兹等人。 [18] 发现掺杂 Sn 4+ 促进了TiO2从锐钛矿到金红石的相变。
以往的研究主要集中在通过共沉淀法、溶胶-凝胶法和固相反应制备柴油氧化催化剂[5, 6, 15, 19, 20]。杨等人。 [19] 通过共沉淀法制备了 Pt/TiO2 催化剂,发现 CO 和 C3H6 的转化率在 232°C 时仅达到 50%。李等人。 [15] 通过溶胶-凝胶法合成了 TiO2-SnO2 纳米复合材料,并表明 TiO2-SnO2 在 260°C 下转化为 CO 的转化率为 90%。谢里夫等人。 [6]制备的Ru/[Ca24Al28O64] 4+ (O 2− )2 通过固相反应表明 Ru/[Ca24Al28O64] 4+ (O 2− 由于 Ru 的分散性较低,在 240 °C 时,)2 与 CO 的比率仅为 82%。因此,柴油氧化催化剂的低温活性仍然存在关键挑战,仍然需要大量努力来去除柴油冷启动中产生的 CO 和 HC。此外,目前的研究 [8, 16, 19, 21, 22] 主要集中在共沉淀法和溶胶-凝胶法制备 DOC 催化剂,其粒径小,但样品结晶度差,多重晶相;此外,还需要通过共沉淀法对混合物进行后续的热处理工艺。在制备过程中采用水热处理,避免了传统的煅烧过程和催化剂的硬聚集的形成,可以提高低温催化活性[23]。但目前对一步法水热法缺乏系统全面的研究[24, 25]。
因此,我们报道了 RuOx Sn 4+ 上支持的粒子 一步水热法改性的 TiO2 是优异的 CO 和 HC 氧化催化剂,具有良好的低温活性和稳定性。一系列 Snx Ti1−x 氧气 (x =0, 0.33, 0.5, 0.67, 1) 固溶体采用一步水热法制备。 Ru/Snx Ti1−x 然后通过浸渍 Snx 制备 O2 Ti1−x O2 与 RuCl3 一起氧化 CO 和 C3H8。水热温度、水热时间、煅烧温度和Ru/Snx的Sn/Ti摩尔比的影响 Ti1−x 研究了O2催化剂以提高低温活性和稳定性。
方法
材料
五水氯化锡(IV)(SnCl4·5H2O)购自广东科华股份公司,钛酸四丁酯(C16H36O4Ti)购自天津科米欧化学试剂厂,无水氯化钌(III)RuCl3(37% Ru w/w) ) 购自阿拉丁。
催化剂的制备
Snx Ti1−x O2固溶体采用一步水热法制备。将一定量的SnCl4·5H2O和C16H36O4Ti分别溶于200 mL去离子水和10 mL无水乙醇中;然后,将C16H36O4Ti乙醇溶液和SnCl4·5H2O水溶液混合,在室温下搅拌0.5 h。将均匀混合物放入 250 mL 高压釜中,在 180 °C 下保持 24 h。之后,将混合溶液用去离子水和乙醇离心洗涤数次,直至无Cl - 残留 ,然后在烘箱中在 80°C 下干燥过夜。随后得到浅黄色固体产物,命名为Snx Ti1−x 氧气。 SnO2和TiO2分别采用相似的制备方法得到。
Ru/Snx Ti1−x Snx浸渍制备O2催化剂 Ti1−x 含 1.0 wt.% RuCl3 的水溶液的 O2。将这些样品超声搅拌2 小时,在80℃下干燥12 小时,然后在400℃下煅烧3 小时(升温速率为3℃/分钟)。所得粉末命名为Ru/Snx Ti1−x 氧气。
催化性能
在带有电加热器的固定床石英反应器上评估催化剂的活性。模拟反应气体包含 3000 ppm CO、600 ppm C3H8、600 ppm NO、50 ppm SO2、7% O2 和 N2 的混合物,气体空速为 60,000 mL g -1 h −1 .气体流速由质量流量控制器调节。固定床的温度通过放置在中心通道中间的0.5-mm K-热电偶测试。出口 CO 和 C3H8 由 KM9106 烟气分析仪(Kane International Limited,英国)测量。转换 (X ) CO 和 C3H8 的计算公式如下:
$$ X=\frac{c_{\mathrm{in}}-{c}_{\mathrm{out}}}{c_{\mathrm{in}}}\times 100\% $$其中 c in 是 CO 或 C3H8 和 c 的初始浓度 out 是反应温度下 CO 或 C3H8 的瞬时值; T 50表示为低温催化活性指数。
催化剂表征
样品的 X 射线衍射 (XRD) 图案在配备有使用 Cu Kα 辐射 (0.15418 nm) 的高温室的 BRUKER D8 ADVANCE 衍射仪上通过功率 X 射线衍射进行。 X射线管工作在40 kV×40 mA的源功率下。
Brunauer-Emmett-Teller (BET) 表面积通过在 Micromeritics ASAP2020 吸附装置上在 77 K 下进行氮吸附测试;比表面积和孔分布分别通过 BET 和 BJH 方法计算。每次分析前,这些样品在300°C下真空脱气4 h。
使用 Nicolet is5 光谱仪以 4.0 cm -1 的光谱分辨率检查傅里叶变换红外 (FT-IR) 光谱 .将粉末压成自支撑晶片(约 15 mg,12 mm 直径)。晶片在 300°C 下用 N2 预处理 1 h。冷却至室温后,记录样品光谱。
这些样品的透射电子显微镜 (TEM) 图像是通过 Tecnai G2 F20 仪器在 200 kV 的加速电压下获得的。观察前将样品研磨、分散于乙醇中并沉积在碳包覆铜网上。
X射线光电子能谱(XPS)分析在ESCALAB250Xi光谱仪上进行,使用单色Al Kα辐射(1486.6 eV),加速功率为15 kW。所得样品光谱以C1s(284.6 eV)为内参进行校正。
H2-程序升温还原 (H2-TPR) 实验在连接到热导检测器 (TCD) 的石英反应器中进行,其中 H2 (6.9% vol. %)-Ar 混合物 (30 mL/min) 作为还原剂。在反应之前,将样品 (50 mg) 在 N2 中在 300°C 下预处理 1 小时,然后冷却至室温。 TPR以10℃/min的速率从室温开始到目标温度。
使用与 H2-TPR 相同的设备进行程序升温氧解吸 (O2-TPD) 实验。用过的催化剂 (50 mg) 在 30 mL/min 的流动 Ar 下在 300°C 下预处理 1 小时。然后,在 O2-Ar 混合物(20% O2 vol.%)下在 500 °C 下进行氧吸附 0.5 h。冷却至室温后,将系统在 Ar (30 mL/min) 中吹扫 1 小时。处理后升温至目标温度(10℃/min)。
在 Nicolet 5700 FT-IR 光谱仪上收集 CO 吸附的原位红外光谱 (IR),光谱分辨率为 4.0 cm -1 .通过暴露催化剂的自支撑晶片(约 15 mg)并安装在商业受控环境室(HTC-3)中来进行 CO 吸附。将样品以 5.0 mL/min 的速率暴露于受控的 CO-Ar(10% 体积的 CO)流中 40 分钟。在不同目标温度下以10°C/min的速率从室温到300°C记录光谱。
结果与讨论
催化活性和稳定性
图1显示了CO和C3H8氧化对Ru/Snx的催化活性 Ti1−x O2催化剂在水热温度180℃、水热时间24 h、煅烧温度400℃的最佳制备条件下(图S1、S2和S3)。可以看出,Ru/Snx 的催化性能 Ti1−x O2催化剂随着反应温度的升高先增加后趋于稳定。当Sn/Ti的摩尔比为2/1时,T 50 的 Ru/Sn0.67Ti0.33O2 氧化 CO 和 C3H8 的反应温度分别为 180 °C 和 320 °C,比其他 Sn/Ti 摩尔比低。 CO的转化率在240°C时达到90%,在Ru/Sn0.67Ti0.33O2催化剂上,在500°C时可以实现C3H8的完全转化。每个样品的催化性能相对于表面上的 Ru 原子进行归一化,并以周转频率 (TOF) 表示,如图 2 所示。Ru/Sn0.67Ti0.33O2 的 TOF 值是最高的任何反应温度下的所有样品。这归因于 Sn0.67Ti0.33O2 表面高度分散的 Ru,活性成分 Ru 与载体 Sn0.67Ti0.33O2 有很强的相互作用 [22, 26]。谢里夫等人。 [6]报道了Ru/[Ca24Al28O64] 4+ 的转化 (O 2− 在 240 °C 时,)2 与 CO 的比例仅为 82%。村山等人。 [27] 报道称,在 250 °C 时,Au/Nb2O5 和 Au/SiO2 向 CO 的转化率分别为 55% 和 38%。与其他文献 [27, 28] 相比,当 Sn/Ti 的摩尔比为 2/1 时,本研究可以在较低温度下实现更高的 CO 转化率。奥卡尔等人。 [29] 报道了 T 当 Ru 的负载量为 0.5 wt.%、1.0 wt.% 和 4.5 wt.% 时,Ru/ZnAl2O4 催化剂氧化的 CH4 的 50 分别为 480、500 和 540 °C。威尔伯恩等人。 [30] 报道了 T 在 0.3Pd–0.7Pt/γ–Al2O3 催化剂上的 CH4 氧化 50 为 360 °C。不同催化剂对CO和C3H8氧化的催化活性见表S1和表S2。本工作可以在较低温度下实现 C3H8 的完全转化。 Sn/Ti的最佳摩尔比为2/1,这与CO的活性一致。从以上分析可以得出结论,CO和C3H8的转化率受Sn/Ti摩尔比的影响很大。当Sn/Ti的摩尔比为2/1时,T Ru/Sn0.67Ti0.33O2 对 CO 和 C3H8 的 50 分别为 180 °C 和 320 °C。反应温度为240℃时,CO的转化率可达90%,反应温度为500℃时,可实现C3H8的完全转化。
<图片>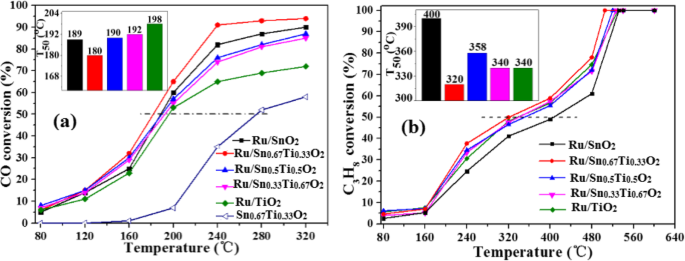
不同Sn/Ti摩尔比对Ru/Snx的影响 Ti1−x O2 催化氧化 CO (a ) 和 C3H8 (b )
<图片>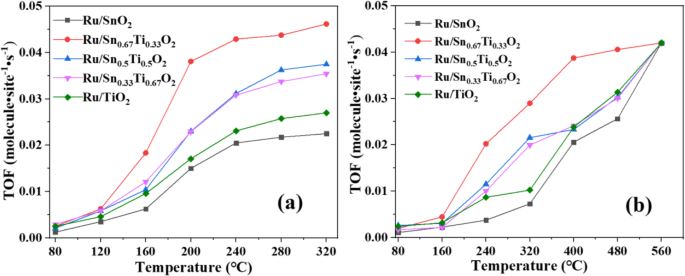
CO 反应温度的 TOF (a ) 和 C3H8 (b ) 在各种催化剂上氧化
在图 3 中研究了 CO 和 C3H8 的稳定性,在 180°C 的水热温度、24 h 的水热时间和 400°C 的煅烧温度下(图 S1、S2 和 S3)。 CO在240℃时转化率达到90%,C3H8在500℃时可完全转化。有趣的是,Ru/Sn0.67Ti0.33O2 催化剂在催化反应 12 h 后基本失活;然而,Ru/TiO2 和 Ru/SnO2 催化剂在氧化 CO 时的活性随着时间的增加而略有下降。这一现象表明 Snx 的形成 Ti1−x O2固溶体不仅可以提高催化剂的活性,还可以增加稳定性。归因于Ru高度分散在Sn0.67Ti0.33O2表面;活性组分Ru与载体Sn0.67Ti0.33O2之间存在强相互作用[26]。
<图片>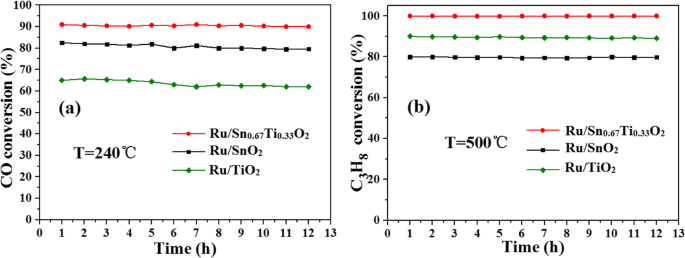
Ru/Snx的稳定性 Ti1−x O2 催化 CO (a ) 和 C3H8 (b )
催化剂表征
Snx 的纹理属性 Ti1−x O2 支持和 Ru/SnxTi1−x 氧气催化剂
图 4 显示了两种 Snx 的 XRD 谱 Ti1−x O2固溶体和Ru/Snx Ti1−x O2 催化剂。在 TiO2 (25.78°) 和 Ru/TiO2 (25.67°) 样品中观察到典型的锐钛矿结构峰,晶粒尺寸分别约为 4 nm 和 5.5 nm(表 1)。随着 Sn 的引入,出现了从锐钛矿到金红石的相变。未观察到Ru衍射峰,表明Ru高度分散在Snx上 Ti1−x O2 表面或超出 XRD 检测限制 [31]。此外,Snx 的衍射峰 Ti1−x O2 和 Ru/Snx Ti1−x 随着 Sn 含量的增加,O2 逐渐向低角度移动,表明晶面间距 d 根据布拉格方程增加,2d sinθ =nλ .这与四方晶格参数(a 和 c ) 在表 1 中,这归因于较大离子半径 Sn 4+ 的取代 (0.071 nm) 对于 Ti 4+ (0.068 nm)。结果表明 Sn 4+ 已成功掺杂到 TiO2 晶格中形成均匀的 (–Sn 4+ –O–Ti 4+ –) 固溶体,同时保持金红石相结构,这与之前的一些研究一致[5, 18]。
<图片>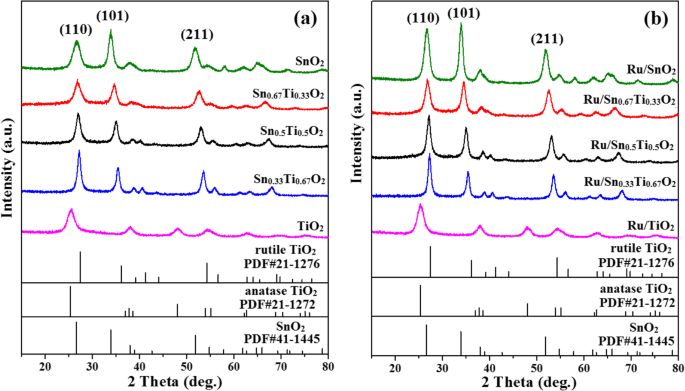
Snx的XRD图谱 Ti1−x O2 支持 (a ) 和 Ru/Snx Ti1−x O2 催化剂 (b )
为了确定样品的质地特性,使用了 N2 吸附-解吸技术。这些样品的N2吸附-解吸等温线和相应的孔径分布曲线如图5所示。SnO2的N2吸附-解吸等温线明显属于II型;其他是根据 IUPAC 分类的经典 IV 型,并在 p/p 中呈现 H2 复合滞后回线 0 0.4-0.95 的范围,这是介孔材料的一个共同特征(图 5a,c)[17, 32]。这些介孔的存在是催化剂比表面积大的重要原因[33]。所有 Snx Ti1−x O2 支持和 Ru/Snx Ti1−x O2 催化剂表现出狭窄的小孔分布(3-8 nm),尤其是 Sn0.67Ti0.33O2 载体和 Ru/Sn0.67Ti0.33O2 催化剂,孔径主要均匀分布在 5 nm 附近(图 5b) , d)。这一现象表明适量的Sn可以减弱催化表面的扩散系数,间接阻碍微晶的团聚[17]。
<图片>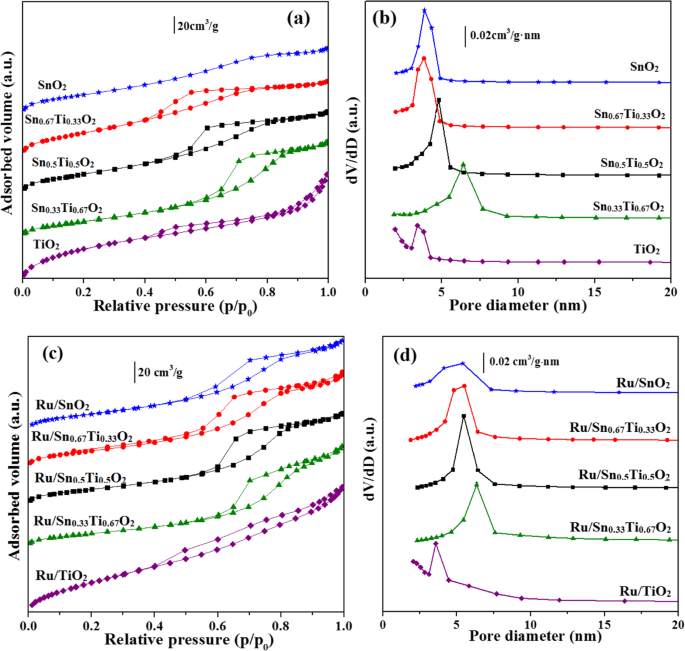
Snx的N2吸附-解吸等温线 Ti1−x 氧气 (a ) 和 Ru/Snx Ti1−x 氧气 (c ) SnxTi1−x 的孔径分布 氧气 (b ) 和 Ru/Snx Ti1−x 氧气 (d )
Snx的织构特性 Ti1−x O2 支持和 Ru/Snx Ti1−x O2催化剂列于表1中。比表面积和孔分布通过BET和BJH方法计算。 Sn0.67Ti0.33O2的比表面积和孔容均为156.5 m 2 g −1 和 0.17 cm 3 g −1 , 分别。但与 Sn0.67Ti0.33O2 载体相比,Ru/Sn0.67Ti0.33O2 催化剂的比表面积和孔体积均降低,这表明 Ru 负载在 Sn0.67Ti0.33O2 表面。此外,在高温煅烧过程中,Ru/Sn0.67Ti0.33O2 催化剂被烧结,开孔结构坍塌形成堵塞的孔[31]。尽管如此,Ru/Sn0.67Ti0.33O2 仍保持较大的比表面积(83.3 m 2 g −1 ) 和较小的孔径 (5.3 nm) 与其他金红石样品如 Ru/Sn0.33Ti0.67O2、Ru/Sn0.5Ti0.5O2 和 Ru/SnO2 相比。
图 6 为 Snx 的 FT-IR 光谱 Ti1−x O2 支持和 Ru/Snx Ti1−x O2 催化剂。所有样品在类似的波数位置都呈现出类似的振动峰。在3223.68 cm −1 附近的吸附 是由于表面羟基邻近氧空位 [34, 35]。 1501.86–1618.18 cm −1 的波段 属于水的角振动峰。晶格氧对称伸缩振动峰出现在1028.17 cm -1 . 527.27–681.2 cm −1 的波段 可能归因于 TiO2 或 SnO2 的伸缩振动峰 [34]。与Snx相比 Ti1−x O2 支持,Ru/Snx Ti1−x O2 光谱变宽,表明活性成分 Ru 和支持 Snx Ti1−x O2 存在一定的相互作用,导致催化剂表面缺陷[36, 37]。
<图片>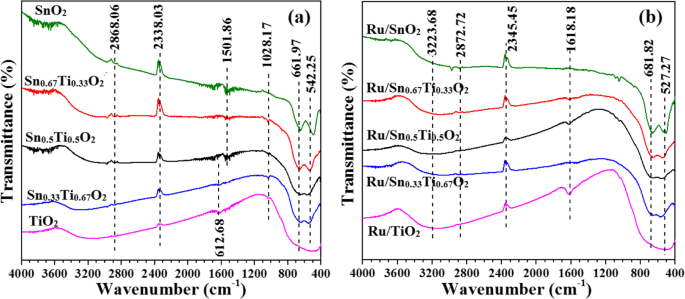
Snx 的 FT-IR 光谱 Ti1−x O2 支持 (a ) 和 Ru/Snx Ti1−x O2 催化剂 (b )
催化剂的形态
低分辨率和高分辨率 TEM、HRTEM 图像以及 Ru/Snx 的粒度分布 Ti1−x O2 如图 7 所示。 根据对图 7a、d、g、j 和 m 中呈现的 TEM 图像的观察,我们发现所有样品均由具有不规则形状和无序介孔结构的明确颗粒组成,这是由纳米颗粒的团聚形成的 [38]。此外,可以看出 Ru/Sn0.67Ti0.33O2 样品具有最高的团聚度,因为这些样品中晶粒尺寸最小。从 HRTEM 图像(图 7b,e,h,k,n)中,只有一种晶格条纹 0.327 nm,与这些样品的(110)面兼容。此外,我们发现没有观察到 TiO2 和 SnO2 的晶格条纹,这归因于 Sn 4+ 已成功掺杂到 TiO2 的晶格中以形成均质的 Snx Ti1−x O2 固溶体[39]。结果与XRD一致。 Ru 颗粒尺寸分布(图 7c、f、i、l、o)表明 Ru 颗粒的近似尺寸范围为 3 到 20 nm。 Sn 4+ 的介绍 可以有效地减小 Ru 颗粒的尺寸并在 Snx 上实现更高的分散 Ti1−x O2 表面。与其他样品相比,Ru/Sn0.5Ti0.5O2 样品的 Ru 粒径分布更宽(<13 nm),这可能是由于 (–Sn 4+ –O–Ti 4+ –) 物种和 Ru [26]。 Ru/Sn0.67Ti0.33O2催化剂在所有样品中具有更好的Ru分散性和更小的粒径(5.49 nm)。
<图片>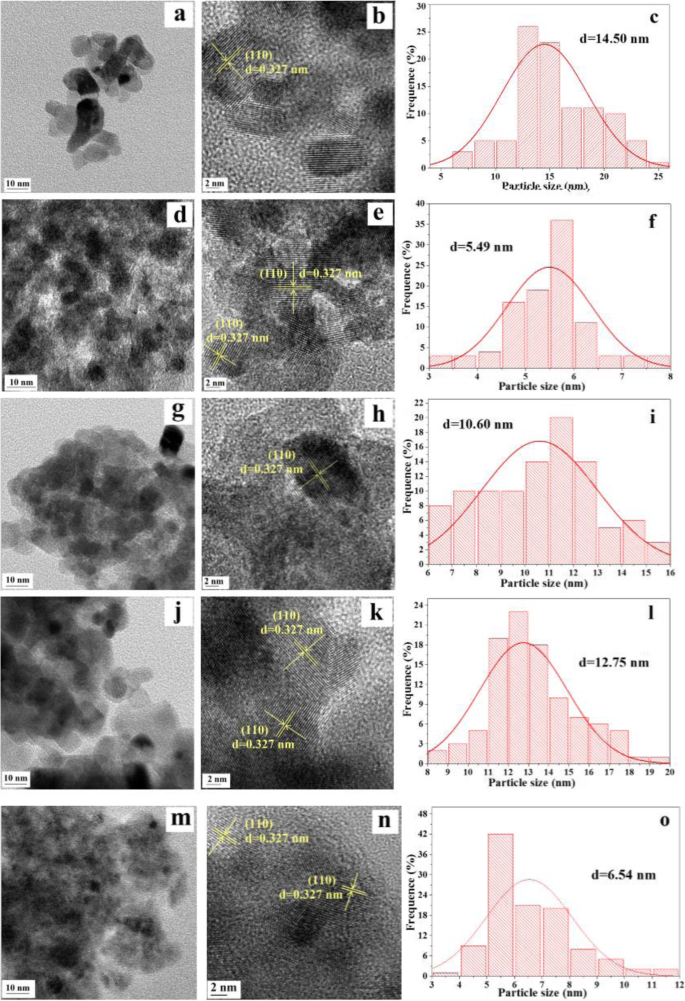
TEM、HRTEM 图像和 a 的粒径分布 , b , c Ru/SnO2; d , e , f Ru/Sn0.67Ti0.33O2; g , h , i Ru/Sn0.5Ti0.5O2; j , k , l Ru/Sn0.33Ti0.67O2;和 m , n , o Ru/TiO2
催化剂的表面特性
为了进一步确定元素状态和表面组成,进行了 XPS 分析。图 8 显示了 Snx 的 Sn 3d、Ti 2p、O 1s 和 Ru 3d 的 XPS 光谱 Ti1−x O2 支持和 Ru/Snx Ti1−x O2 催化剂。 Sn 3d3/2 和 Sn 3d5/2 的 XPS 结合能值分别在 486.6-487.5 eV 和 494.9-496.1 eV 处观察到,这是 Sn 4+ 的特征 Snx 中的物种 Ti1−x O2 支持或 Ru/Snx Ti1−x O2 催化剂。有趣的是,在引入 Sn 4+ 后,Sn 3d3/2 和 Sn 3d5/2 的结合能转移到更高的值 , 表示部分 Sn 4+ 替换 Ti 4+ 位点并与 TiO2 有很强的相互作用,这与 XRD 一致。此外,氧空位可能由较低价的 Sn δ+ 产生 [5]。在 Ti 2p 的 XPS 光谱中在 458.7–459.9 eV 和 464.3–465.8 eV 处观察到两个对应于 Ti 2p3/2 和 Ti 2p1/2 的峰,表明 Ti 4+ 和 Ti 3+ 样品中存在,Ti 2p3/2 和Ti 2p1/2 的结合能值随着Sn 4+ 的增加而向更高的结合能值移动 ,进一步证明氧空位的存在。从表 3 可以看出,XPS 观察到的 Sn/Ti 摩尔比略高于理论计算值,表明 Sn 在催化剂表面富集,导致更多的氧空位。由于Sn的电负性(1.96)大于Ti的电负性(1.62),换言之,Sn的电子俘获能力强于Ti,导致氧化还原平衡(Sn 4+ +Ti 3+ → Sn δ+ +Ti 4+ ) 向右移动 [32]。
<图片>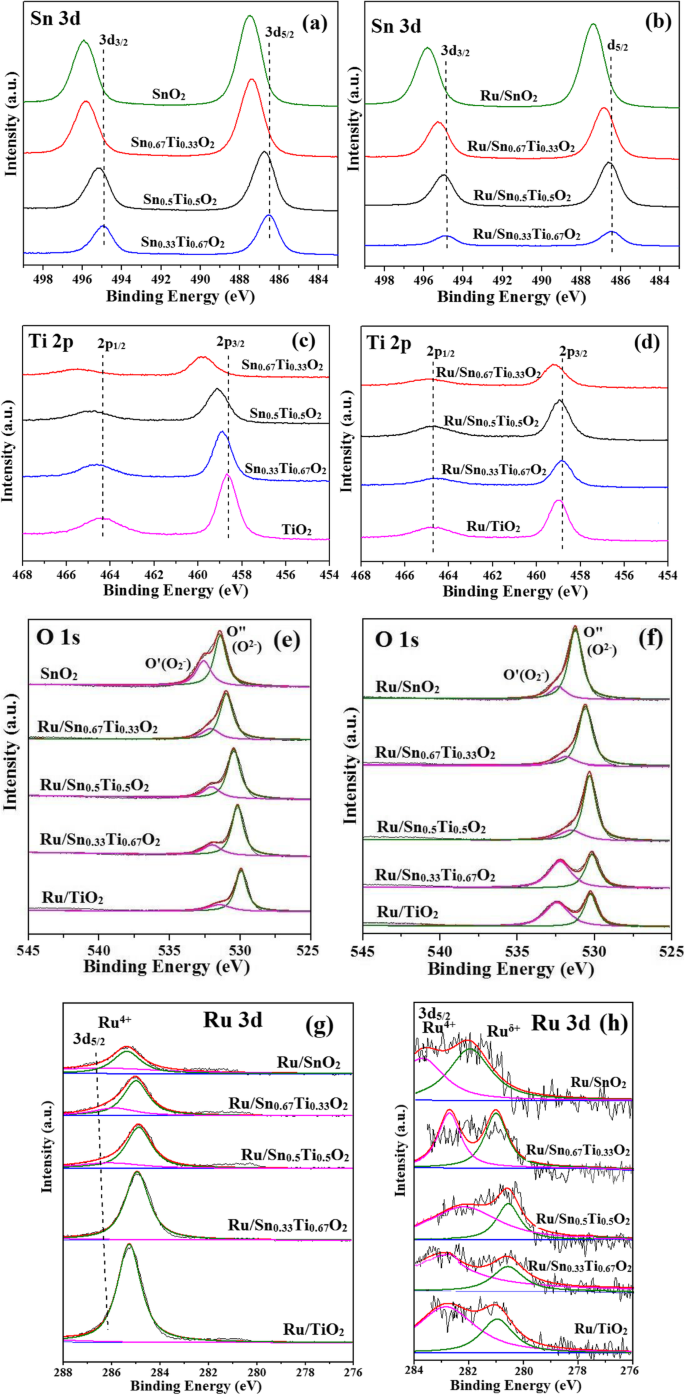
XPS 光谱 (Sn3d (a , b ), Ti2p (c , d ), O1s (e , f ) 和 Ru3d (g , h )) 的 Snx Ti1−x O2 支持和 Ru/Snx Ti1−x O2催化剂
O 1s 电离特征的高分辨率光谱在数值上与高斯特征一致,并解卷积为两个峰 [5]。较高的结合能 (O', 532.1 eV) 是由形成 (O2 − , O − , 或 O2 2− ) 物种。然而,O” (529.9 eV) 是 O 2− 的特征峰 金属氧化物表面。有趣的是,添加 Sn 4+ 后,O 1s 的结合能转移到更高的值 .
Ru 3d 光谱呈现 Ru 4+ 和较低的值 Ru δ+ . Ru 3d5/2 的信号通常用于分析 Ru 物种的电荷状态,因为另一个 Ru 3d3/2 在 284.0 eV 附近与 C 1s 重叠 [40]。 282.0–283.5 eV的结合能分配给Ru 3d5/2,对应于Ru 4+ .大约 280.2–281.7 eV 的较低结合能归因于较低态的 Ru δ+ , 和 Ru δ+ Ru/Sn0.67Ti0.33O2的相对比达到53.9%,高于其他催化剂。可以解释为Sn0.67Ti0.33O2与Ru之间的强相互作用导致了大量的表面活性氧[26]。
进行 XPS 和 EDS 分析以确定样品的表面和本体组成,如表 2 所示。表面和本体 Ru 分析表明 Ru/Sn0.67Ti0.33O2 具有最高的表面 Ru (0.69 wt.%) 和本体 Ru (0.40 wt.%) 在所有催化剂中,表明活性组分 Ru 更均匀地分布在 Sn0.67Ti0.33O2 载体上,更多的 Ru 物种进入 Sn0.67Ti0.33O2 内部形成强相互作用。
为了进一步研究 Ru/Snx 的还原性能 Ti1−x 进行 O2 催化剂、程序升温还原研究(图 9)。这些 H2-TPR 剖面的形状几乎相同。 Ru/Snx的还原峰 Ti1−x O2 分为两部分:80-270 °C 的低温还原峰与低态 Ru δ+ 相关 由 RuO2 和大量 Sn 4+ 还原 可还原为低价Sn δ+ 或可归因于表面氧的还原 [41],而 600–640 °C 的高温还原峰与 Sn 0 相关 从 Sn δ+ 减少 or the reduction of bulk oxygen of catalysts [26, 42], which is consistent with XPS results. The reduction temperature of Ru/Snx Ti1−x O2 moves towards lower temperature, peaks broaden and H2 consumption increase with the addition of Sn, and hydrogen consumption from the H2-TPR measurements are shown in Table 3. The dispersion of active components on the surface of the samples has a significant effect on the reduction of surface oxygen, and hydrogen could be more easily activated with higher dispersion of Pd, resulting in the increase of H2 consumption [43]. Therefore, we can infer that the introduction of Sn significantly increased the dispersion of Ru on the carrier, which may have resulted from the formation of Snx Ti1−x O2 solid solution. The results are in good agreement with XRD and TEM. Because the reduction of TiO2 is usually difficult to conduct at low temperature, there are no peaks of the TiO2 reduction observed during the H2-TPR from 50 to 800 °C [15]. Nevertheless, the Ru/Sn0.67Ti0.33O2 still exhibits a higher H2 consumption.

H2-TPR spectra of Ru/Snx Ti1−x O2 catalysts
The O2-TPD experiments (Fig. 10) of Ru/Snx Ti1−x O2 samples are imposed to gain insight into the mobility of surface and lattice oxygen. The signal at low temperature (<200 °C) is attracted by the desorption of surface chemisorbed oxygen (O2 − , O2 2− , or O − species); the main peak centered at 280 °C or 500 °C which is attributed to the desorption of the structure oxygen species, and the peaks above 600 °C are assignable to the desorption of the lattice oxygen (O 2− ) species [44]. The incorporation of Sn increased the adsorbed oxygen species and shifted to a lower temperature [45]. The results indicate that the incorporation of Sn improved the oxygen activation ability of the Ru/Snx Ti1−x O2 samples and the interaction between the carriers Snx Ti1−x O2 and active component Ru [46, 47].

O2-TPD spectra of Ru/TiO2 (a ), Ru/Sn0.33Ti0.67O2 (b ), Ru/Sn0.5Ti0.5O2 (c ), Ru/Sn0.67Ti0.33O2 (d ), and Ru/SnO2 catalysis
CO and/or O2 Interaction with these Samples
The in situ FI-IR spectra of CO adsorption are recorded to further investigate the effect of the ruthenium oxide species, as shown in Fig. 11. The band located at 2052 cm −1 is attributed to linear CO adsorbed on reduced Ru crystallites (Ru δ+ –CO), the band at 2140 cm −1 and 2075 cm −1 can be assigned to two different types of multicarbonyl species on partially oxidized Ru sites (Ru n+ (CO)x ), and the band at 1765 cm −1 is attributed to (Snx Ti1−x O2)Ru–CO species [48, 49]. The Ru δ+ –CO adsorption peaks at room temperature indicate the presence of some lower state Ru δ+ 物种。 This is in agreement with the XPS results. However, the desorption temperature of the Ru δ+ –CO peak is related to the Sn/Ti ratio and temperature. As the temperature increases, the peak intensity enhances firstly and then decreases gradually. Simultaneously, the CO adsorption peak moves to a higher wave number (2052 cm −1 at 25 °C and 2060 cm −1 at higher temperatures). This red-shift indicates that Sn 4+ has stronger electron-donating capability [50]. For the Ru/SnO2, Ru/Sn0.5Ti0.5O2, Ru/Sn0.33Ti0.67O2, and Ru/TiO2 samples, the CO maximum adsorption peak on Ru δ+ appears at about 200 °C and disappears basically at 300 °C. For the Ru/Sn0.67Ti0.33O2 sample, the CO maximum adsorption peak on Ru δ+ appears at about 200 °C, which can be observed clearly even at 300 °C. It can be concluded that Ru δ+ is much more stable in Ru/Sn0.67Ti0.33O2 sample, which can provide more CO adsorption sites than in the other samples.

In situ FI-IR spectra of the 10% CO/Ar interaction with a Ru/SnO2, b Ru/Sn0.67Ti0.33O2, c Ru/Sn0.5Ti0.5O2, d Ru/Sn0.33Ti0.67O2, e Ru/TiO2 at different temperatures
Possible Reaction Mechanism over the Ru/Snx Ti1−x O2 Catalysts
According to the characterizations mentioned above, a possible reaction mechanism of CO and C3H8 oxidation is proposed and schematized in Fig. 12. Based on the XPS results, electrons migrate between Ru and Snx Ti1−x O2 solid solution; because the electronegativity of Ru (2.22) is larger than that of Ti (1.62) and Sn (1.96), the electrons will transfer from the Snx Ti1−x O2 solid solution to Ru 4+ , in which lower state Ru δ+ will be generated. Meanwhile, –Ti 4+ –O–Sn 4+ – species are oxidized and more oxygen will be absorbed on the surface of Snx Ti1−x O2 solid solution, which can provide oxygen to the oxidation reaction of CO and C3H8. At the same time, the by-products produced in the oxidation process will also be adsorbed on the surface of Snx Ti1−x O2 solid solution, which will not deteriorate the activity of Ru δ+ 物种。 It is also the reason for the high stability of the catalysts. Moreover, the lower state Ru δ+ species have more metal properties, which play a crucial role in the activation of CO and C3H8 [40]. Compared with Ru/TiO2 and Ru/SnO2, high dispersion of Ru on Snx Ti1−x O2 solid solution is also an important cause for their excellent activity and stability. Based on O2-TPD analysis, O2 is first adsorbed on the surface of catalysts to form O2 − species and CO and C3H8 adsorbed on Ru δ+ species react with O2 − species to produce CO2 and H2O, which is a Langmuir-Hinshelwood mechanism.

Possible reaction mechanism of CO and C3H8 over Ru/Snx Ti1−x O2
结论
A series of Ru/Snx Ti1−x O2 catalysts were prepared by a one-step hydrothermal method for the catalytic oxidation of CO and C3H8. The preparation conditions of Ru/Snx Ti1−x O2 catalysts were optimized for CO oxidation reaction. Ru/Sn0.67Ti0.33O2 catalyst shows best CO catalytic activity and stability at low temperature under the condition of hydrothermal temperature at 180 °C, hydrothermal time at 24 h, and calcination temperature at 400 °C.
The effects of different molar ratios of Sn/Ti on the catalytic properties of Ru/Snx Ti1−x O2 catalysts for CO and C3H8 were investigated under the optimum preparation conditions. The results show that the Ru/Sn0.67Ti0.33O2 catalyst exhibits better low-temperature activity and stability. The conversion of CO reached 90% at 240 °C, and T 50 of which keeps at 180 °C. The complete conversion of C3H8 could be achieved at 500 °C, and its T 50 remains at 320 °C. The excellent catalytic activity of Ru/Sn0.67Ti0.33O2 catalyst is attributed to the factors listed as follows.
- (1)
The successful incorporation of Sn 4+ into the TiO2 lattice to replace Ti 4+ forms a homogeneous solid solution (–Sn 4+ –O–Ti 4+ – species), which enhances the interaction between active component Ru and carrier Snx Ti1−x 氧气。 The crystal growth of the anatase phase can be inhibited by the introduction of Sn 4+ , which results in the presence of the rutile phase.
- (2)
Ultrafine Ru nanoparticles (~ 5 nm) are highly dispersed on Snx Ti1−x O2 support, suggesting that the introduction of Sn 4+ could not only prevent grain agglomeration and induce a smaller grain size, but also produce more defects such as oxygen vacancies.
- (3)
CO and C3H8 species can be absorbed on Ru δ+ sites; O2 − is formed by the adsorption of O2 on the oxygen vacancies. The adsorbed CO and C3H8 react with O2 − to produce CO2 and H2O.
数据和材料的可用性
All data generated or analyzed during this study are included in this published article and supporting information.
缩写
- XRD:
-
X射线衍射
- 赌注:
-
Brunauer-Emmett-Teller
- FT-IR:
-
傅里叶变换红外
- TEM:
-
透射电子显微镜
- XPS:
-
X射线光电子能谱
- H2-TPR:
-
H2-temperature-programmed reduction
- O2-TPD:
-
Temperature-programmed oxygen desorption
- DOC:
-
Diesel oxidation catalysts
- SCR:
-
Selective catalytic reduction
- DPF:
-
Diesel particulate filter
- SOF:
-
Soluble organic fraction
纳米材料
- 钴掺杂 FeMn2O4 尖晶石纳米粒子的制备和磁性
- 走向 TiO2 纳米流体——第 1 部分:制备和性质
- SrTiO3 改性金红石型 TiO2 纳米纤维的一步静电纺丝路线及其光催化性能
- Sb/坡缕石 (PAL) 纳米颗粒的制备和增强催化氢化活性
- Li/Nb 比对 Li-Nb-O 化合物制备和光催化性能的影响
- Au@TiO2 蛋黄-壳纳米结构的制备及其在亚甲基蓝降解和检测中的应用
- 中空结构LiNb3O8光催化剂的制备和光催化性能
- PPy 包覆的 MnO2 混合微材料的制备及其作为锂离子电池阳极的改进循环性能
- 具有增强光催化性能的新型 Bi4Ti3O12/Ag3PO4 异质结光催化剂
- 钯(II)离子印迹聚合物纳米球的制备及其从水溶液中去除钯(II)
- 一维混合二元氧化物 CeO2-LaO x 支持的金催化剂的合成和 CO 氧化活性
- ITO/PtRh:PtRh 薄膜热电偶的制备和热电特性